The Interactive Fly
Genes involved in tissue and organ development
Cytochrome C has two apparently separable cellular functions: respiration and caspase activation during apoptosis. While a role of the mitochondria and cytochrome C in the assembly of the apoptosome and caspase activation has been established for mammalian cells, the existence of a comparable function for cytochrome C in invertebrates remains controversial. Drosophila possesses two cytochrome c genes, cyt-c-d and cyt-c-p. Only cyt-c-d is required for caspase activation in an apoptosis-like process during spermatid differentiation, whereas cyt-c-p is required for respiration in the soma. However, both cytochrome C proteins can function interchangeably in respiration and caspase activation, and the difference in their genetic requirements can be attributed to differential expression in the soma and testes. Furthermore, orthologues of the apoptosome components, Ark (Apaf-1) and Dronc (caspase-9), are also required for the proper removal of bulk cytoplasm during spermatogenesis. Finally, several mutants that block caspase activation during spermatogenesis were isolated in a genetic screen, including mutants with defects in spermatid mitochondrial organization. These observations establish a role for the mitochondria in caspase activation during spermatogenesis (Arama, 2006).
Apoptosis is a morphologically distinct form of active cellular suicide that serves to eliminate unwanted and potentially dangerous cells. The key enzymes responsible for the execution of apoptosis are an evolutionarily conserved family of cysteine proteases known as caspases. Caspases are present in an inactive or weakly active state in virtually all cells of higher metazoans, and their activity is carefully regulated by both activators and inhibitors. In vertebrates, the mitochondria play an important role in the control of apoptosis: they release cytochrome C and other pro-apoptotic proteins in response to various death signals. In the cytosol, cytochrome C binds to Apaf-1 (Zou, 1997) which in turn promotes the assembly of a multiprotein complex, termed the 'apoptosome', and caspase-9 activation (Rodriguez, 1999; Adams, 2002; Cain, 2002; Salvesen, 2002). In the ensuing 'caspase cascade', many intracellular substrates are cleaved and apoptosis is executed. However, the exact physiological role of cytochrome C for caspase activation remains to be determined, and a recent report on a mutant cytochrome c that fails to activate Apaf-1 in the mouse (Hao, 2005) suggests that cytochrome C is required for caspase activation in only some mammalian cell types (Arama, 2006).
In invertebrates, any role of cytochrome C for the activation of caspases has remained highly controversial. Whereas RNAi experiments in Drosophila S2 cells have failed to reveal a role for cytochrome C in apoptosis, other reports suggest that cytochrome C may promote caspase activation (Dorstyn, 2002, 2004; Zimmermann, 2002). Drosophila contains two Apaf-1 isoforms: one with a WD40 repeat domain, the target for cytochrome C binding, and another lacking this domain, similar to Caenorhabditis elegans Ced-4. The large isoform can directly bind cytochrome C in vitro and promote cytochrome C-dependent caspase activation in lysates from developing embryos (Kanuka, 1999). Furthermore, an overt alteration in the cytochrome C immuno-staining can be detected in doomed cells in some Drosophila tissues, and the mitochondria from apoptotic cells can activate cytosolic caspases (Varkey, 1999). Finally, disruption of one of the two Drosophila cytochrome c genes, cyt-c-d, is associated with a failure to activate caspases in an apoptosis-like process during sperm terminal differentiation in Drosophila (Arama, 2003). In this process, also known as spermatid individualization, the majority of cytoplasm and cellular organelles are eliminated from the developing spermatids in an apoptosis-like process that requires caspase activity (Arama, 2003). However, it was suggested that the mutants used in s previous study (Huh, 2004) may also affect other genes located in the vicinity of the cyt-c-d locus (Arama, 2006).
In order to rigorously address this issue, a series of genetic and transgenic rescue experiments were conducted that unequivocally establish a role of cytochrome C for caspase activation during Drosophila spermatogenesis. First, a point mutation was isolated in cyt-c-d that is defective in caspase activation. Next, it was demonstrated that transgenic expression of cyt-c-d restores effector caspase activation and rescues all the sterility phenotypes associated with various cyt-c-d mutant alleles. The possibility that cyt-c-p functions specifically in respiration was investigated, whereas cyt-c-d plays a role in caspase regulation. Surprisingly, it was found that expression of either cyt-c-d or cyt-c-p can restore caspase activation in cyt-c-d-deficient spermatids, demonstrating that both proteins are functionally equivalent. Other apoptosome proteins in Drosophila, Ark (Apaf-1) and Dronc (caspase-9) are also required for spermatid individualization, and their mutant phenotypes are similar to spermatids with a block in caspase activity. Surprisingly, however, some active caspase-3 staining can still be detected in these mutant testes, suggesting that cytochrome-C-d may function in yet other unknown pathways to promote caspase-3 activation. Finally, several mutants affecting spermatid mitochondria were identifed that provide a strong link between mitochondrial organization and caspase activation during sperm development (Arama, 2006).
In mammals, mitochondria are important for the regulation of apoptosis, and it has been shown that they can release several proapoptotic proteins into the cytosol in response to apoptotic stimuli. The best-studied case is the release of cytochrome C, an essential component of the respiratory chain. Cytosolic cytochrome C can bind to and activate Apaf-1, which in turn leads to the activation of caspase-9. However, no comparable role of mitochondrial factors for caspase activation has yet been established in invertebrates. The elimination of cytoplasm during terminal differentiation of spermatids in Drosophila involves an apoptosis-like process that requires caspase activity; a P-element insertion (bln1) in one of the two Drosophila cytochrome c genes, cyt-c-d, has been shown to be associated with male-sterility and loss of effector caspase activation during spermatid individualization. This study demonstrates that the defects in caspase activation and spermatid individualization of bln1 mutant males can be rescued by transgenic expression of the ORF of cyt-c-d. Furthermore, from screening more than a thousand male-sterile lines with defects in sperm individualization for defects in active-caspase (CM1) staining, a nonsense point mutation was identified in cyt-c-d, that recapitulates all the phenotypes observed for bln1. Taken together, these results unequivocally demonstrate that cyt-c-d is necessary for effector caspase activation and sperm terminal differentiation in Drosophila (Arama, 2006).
Two decades ago, the mouse cytochrome c gene was used as a probe for screening a Drosophila genomic library and a fragment was isolated that carried two distinct cytochrome c genes. Northern blot analyses indicated high levels of cyt-c-p expression, while cyt-c-d was reported to be expressed at much lower levels in all stages of development. However, neither the exon/intron organization nor the boundaries of the 5' and 3' UTRs of these genes were determined at the time. As a result, the original Northern analyses were performed with a probe corresponding to the untranscribed genomic region between the two cytochrome c genes that was not suitable to properly assess the size and distribution of cytochrome c transcripts. Unfortunately, this has caused considerable confusion in the field from the start, as even the original report noted that the size of the observed cyt-c-d transcript differed more than two-fold from the predicted size. More recently, relying on the incorrect assumption that cyt-c-d is ubiquitously expressed in the fly, it has been suggested that a loss-of-function mutation in cyt-c-d should lead to severe developmental defects and lethality rather than merely male sterility. However, using a specific cyt-c-d 3' UTR probe reveals a transcript of the predicted size that is absent in cyt-c-dbln1 mutants. Furthermore, the RT-PCR and immunofluorescence analyses presented in this study indicate that cyt-c-d is mainly expressed in the male germ line and is completely absent during embryonic and larval development, while cyt-c-p is expressed in the soma during all stages of development. In light of these findings, it is not surprising that loss-of-function mutations in cyt-c-d cause male sterility, whereas cyt-c-p mutations lead to embryonic lethality. RT-PCR results suggest that cyt-c-p is also expressed in the testis, although to a much lower extent than cyt-c-d. This expression is attributed primarily to the somatic cells of the testis, since no cytochrome C protein is detected in cyt-c-dbln1 elongating spermatids, while cyt-c-p RNA is expressed in cyt-c-dbln1 mutant flies. However, the very low cyt-c-d expression detected in the soma of adult females leaves room for the possibility that cyt-c-d might function in caspase activation in some somatic cells as well (Arama, 2006).
In mammalian cells, release of cytochrome C into the cytosol in response to proapoptotic stimuli can be readily demonstrated. However, previous attempts to detect a similar phenomenon in Drosophila have been unsuccessful. In contrast, apoptotic stimuli can lead to increased cytochrome C immuno-reactivity. A possible limitation is that all these studies were conducted using mammalian antibodies with questionable specificity and sensitivity, and only in a small number of cell types and paradigms. Using an antibody that was raised against Drosophila cytochrome C-d, an increase in a 'grainy signal' was detected upon the onset of individualization, with the highest staining observed in the vicinity of the individualization comple (IC). Since it is highly unlikely that additional cytochrome C-d is being transcribed and imported to the mitochondria at this late stage, the explanation is favored that a conformational change or an exposure of a hidden epitope causes the increase in the intensity of the signal. The activation of Dronc, the Drosophila caspase-9 orthologue, also occurs in association with the IC and depends on the presence of the Drosophila Apaf-1 orthologue, Ark. Moreover, the proapoptotic Hid protein is localized in a similar fashion. What are these structures then, which accumulate apoptotic factors in the vicinity of the IC? One plausible suggestion from the literature is that these structures correspond to 'mitochondrial whorls', which result from the extrusion of material from the minor mitochondrial derivative and constitute the leading component of the IC. These 'whorls' can be labeled using a testes-specific mitochondrial-expressed GFP line. Using this GFP marker, it was found that cytochrome C-d is indeed closely associated with mitochondrial whorls. Therefore, it is possible that an active apoptosome forms in the vicinity of the IC in response to dramatic changes in the mitochondrial architecture that occur at this stage of spermatid differentiation. Similarly, studying the response of Drosophila flight muscle cells to oxygen stress, have recently reported that the cristae within individual mitochondria become locally rearranged in a pattern that they termed a 'swirl'. This process was associated with widespread apoptotic cell death in the flight muscle, which was correlated with a conformational change of cytochrome C manifested by the display of an otherwise hidden epitope. Collectively, these observations suggest that apoptosome-like complexes composed of cytochrome C-d, Ark, and Dronc might be associated with unique mitochondrial swirl-like structures. Consistent with this idea, it was found that the long isoform of Ark that contains the WD40 repeats, the target for cytochrome C binding to mammalian Apaf-1, is the major form detectably expressed in testes (Arama, 2006).
The fact that cytochrome C-d immunoreactivity increases in the vicinity of the IC suggests that the extensive mitochondrial organizations preceding individualization may be partially required for caspase activation. Consistent with this idea, several mutants, such as plnZ2-0516, which display defects in Nebenkern differentiation and caspase activation. However, not all mitochondrial differentiation events are required for caspase activation. For example, CM1 staining is seen in fuzzy onions, a mutant defective in the mitochondrial fusion event that generates the Nebenkern. In contrast, analysis of the pln mutant indicates that proper elongation of the Nebenkern is essential for caspase activation. Therefore, characterization of other mitochondrial mutants may shed light on the connection between mitochondrial organization and caspase activation during sperm differentiation (Arama, 2006).
What are the mechanisms by which cytochrome C-d activates caspases during late spermatogenesis? In vertebrate cells, following its release into the cytosol, cytochrome C binds to the WD40 domain of the adaptor molecule Apaf-1, which in turn multimerizes and recruits the initiator caspase, caspase-9 via interaction of their CARD domains. This complex, known as the apoptosome, further cleaves and activates effector caspases like caspase-3. Although this model has become the prevailing dogma in the field, the phenotype of mice mutant for a Cyt c with drastically reduced apoptogenic function ('KA allele') suggests that the mechanisms for caspase activation may be more complex than previously thought. In particular, this study suggests that cytochrome C-independent mechanisms for the activation of Apaf-1 and caspase-9 exist, as well as cytochrome C-dependent but Apaf-1-independent mechanisms for apoptosis. These analyses of ark (Apaf-1) and dronc (caspase-9) loss-of-function mutants demonstrate that both genes are required for spermatid individualization, and that their phenotypes, in particular their failure to properly remove the spermatid cytoplasm into the WB, resemble cyt-c-d mutant spermatids and expression of the caspase inhibitor p35 in the testes. However, some caspase-3-like activity could still be detected in these mutant testes. This may suggest that either the ark and dronc alleles are not null, or that cytochrome C-d also functions in an apoptosome-independent pathway to promote caspase-3 activation. Therefore, the regulation of caspase activation and apoptosis may be more similar between insects and mammals than has been previously appreciated. Further genetic analysis of this pathway in Drosophila may provide general insights into diverse mechanisms of apoptosis activation (Arama, 2006).
Previous observations raised the possibility that the two distinct cytochrome c genes may have evolved to serve distinct functions in respiration and caspase regulation. In order to address this hypothesis, it was asked whether expression of one protein might rescue mutations in the other cytochrome c gene. Surprisingly, it was found that transgenic expression of the cyt-c-p ORF in germ cells rescues caspase activation, spermatid individualization, and sterility of cyt-c-d-/- flies. Therefore, the ability to activate caspases is not restricted to the cytochrome C-d protein, and it is possible that cytochrome C-p functions in apoptosis in at least some somatic cells (Arama, 2006).
Although cyt-c-d is almost exclusively expressed in the male germ cells, ectopic expression of this protein in the soma can rescue the respiration defect and lethality of cyt-c-p-/- mutant flies, demonstrating that cytochrome C-d can function in energy metabolism. This raises the question whether the lack of caspase activation could be due to reduced ATP-levels. Although this is a formal possibility, this explanation is considered very unlikely since mutant spermatids complete many other energy-intensive cellular processes. These include the extensive transformation from round spermatids to 1.8 mm long elongated spermatids, a process that involves extensive remodeling and movement of actin filaments, generation of the axonemal tail, mitochondrial reorganization, plasma/axonemal membranes reorganization, and nuclear condensation and elongation. Since all of these processes can occur in the absence of cytochrome C-d, there is no overt shortage of ATP in cyt-c-d mutants. It is therefore considered very unlikely that ATP has become limiting in these mutant cells. Since earlier stage spermatids express cytochrome C-p, sufficient ATP seems to persist to late developmental stages. In mammalian cells, cellular ATP concentration is sufficiently high (around 2 mM) to keep cultured cell alive for several days upon ATP synthase inhibition. Furthermore, cells in which cytochrome c expression is decreased by RNAi still undergo apoptosis in response to various stimuli. Likewise, it appears that cytochrome C is not essential for the function of mature murine sperm, since mice deficient for the testis specific form of cytochrome C, Cyt cT, are fertile. Taken together, all these observations argue strongly against the possibility that ATP levels in cyt-c-d-/- mutant spermatids would be insufficient for caspase activation (Arama, 2006).
In conclusion, the results presented
in this study definitively demonstrate that cytochrome C-d is essential for caspase
activation and spermatid individualization. Both cytochrome C proteins of
Drosophila are, at least to some extent, functionally interchangeable.
The results also indicate that cytochrome C can promote caspase activation in
the absence of a functional apoptosome. Given the powerful genetic techniques
available, late spermatogenesis of Drosophila promises to be a powerful
system to identify novel pathways for mitochondrial regulation of caspase
activation (Arama, 2006).
The role of mitochondria in Drosophila programmed cell death remains unclear, although certain gene products that regulate cell death seem to be evolutionarily conserved. This study found that developmental programmed cell death stimuli in vivo and multiple apoptotic stimuli ex vivo induce dramatic mitochondrial fragmentation upstream of effector caspase activation, phosphatidylserine exposure, and nuclear condensation in Drosophila cells. Unlike genotoxic stress, a lipid cell death mediator induces an increase in mitochondrial contiguity prior to fragmentation of the mitochondria. Dynamin related protein 1 (Drp1), is important for mitochondrial disruption. Using genetic mutants and RNAi-mediated knockdown of drp-1, it was found that Drp1 not only regulates mitochondrial fission in normal cells, but mediates mitochondrial fragmentation during programmed cell death. Mitochondria in drp-1 mutants fail to fragment, resulting in hyperplasia of tissues in vivo and protection of cells from multiple apoptotic stimuli ex vivo. Thus, mitochondrial remodeling is capable of modifying the propensity of cells to undergo death in Drosophila (Goyal, 2007).
Programmed cell death (PCD) plays an important role in sculpting tissues during animal development. The molecular regulators that are central to this process seem to be evolutionarily conserved from worms to mammals and include autocatalytic initiator caspases, trans-activable effector caspases, cytosolic activating factors (APAF-1), and multidomain Bcl-2 proteins. The proapoptotic Bcl-2-family proteins oligomerize and permeabilize mitochondria, releasing intermembrane space components such as cytochrome-C and Smac/DIABLO into the cytosol, where they activate initiator caspases by an ATP-dependent mechanism. Initiator caspases trans-activate effector caspases that cleave multiple cellular substrates, resulting in DNA degradation, nuclear condensation, and loss of cell integrity (Goyal, 2007 and references therein).
Mitochondrial outer-membrane permeabilization has been proposed to depend on the mitochondrial fission and fusion machinery. Consistent with this, mitochondria undergo dramatic fragmentation very close in time to cytochrome-C release during mammalian cell death. Furthermore, an increase in mitochondrial fragmentation and a block in mitochondrial fusion are essential for cell death progression. In normal cells, the balance in the rates of mitochondrial fission and fusion regulated by Dynamin-related protein-1 (Drp-1), Fis-1 and endophilin (fission), or Mitofusins and Opa-1 (fusion) maintains the dynamic, interconnected mitochondrial tubules. An increase in recruitment of Drp-1 to the mitochondria accentuates staurosporine, lipid, and free oxygen radical stress-induced mitochondrial outer-membrane permeabilization. Moreover, multiple apoptotic stimuli induce mitochondrial recruitment of the proapoptotic Bcl-2-family protein, Bax, to Drp-1 and Mitofusin-2-positive putative mitochondrial fragmentation sites in a Fis-1-dependent manner, consistent with a role for mitochondrial fission and fusion machinery in cell death (Goyal, 2007).
In Drosophila, RHG-family proteins (Reaper, Hid and Grim), genotoxic stresses, and protein synthesis inhibitors antagonize Drosophila inhibitor of apoptosis protein-1 (DIAP-1)-mediated inhibition of the activation of the apical caspase Dronc in an ARK- (Drosophila APAF-1) and ATP-dependent manner, leading to effector caspase activation and cell death. The role of mitochondria in this process is unclear. Cytochrome-C has been shown to be differentially displayed from the mitochondria during cell death. Knockdown of Drosophila cytochrome-C did not affect cell death triggered by genotoxic stress in vitro and ex vivo or developmental stimuli in vivo, although certain nonapoptotic caspase activation pathways utilized during sperm individualization were affected. Furthermore, mitochondrial morphology during Drosophila PCD has not been previously reported (Goyal, 2007 and references therein).
This study shows that multiple apoptotic stimuli result in mitochondrial fragmentation upstream of caspase activation, phosphatidylserine exposure, and nuclear condensation in Drosophila cells. While etoposide induced mitochondrial fragmentation, C6-ceramide resulted in an increase in mitochondrial contiguity prior to its fragmentation. drp-1 mutant or RNAi-treated S2R+ cells are considerably protected from multiple apoptotic stimuli, consistent with reduced mitochondrial fragmentation. Thus, mitochondrial remodeling plays an important role in modifying the propensity of cells to undergo PCD in Drosophila (Goyal, 2007).
Precisely timed ecdysone pulses induce Reaper and Hid expression in the Drosophila larval midgut (0 hr after puparium formation [APF]) or the salivary gland (10 hr APF) and trigger developmental PCD. Mitochondria, visualized by using matrix-targeted GFP (Mito-GFP) in acridine orange-positive, dying prepupal midgut cells (1 hr APF) and salivary glands (minus 4 hr APF), are remarkably fragmented, unlike third-instar larval (-4 hr APF) mitochondria. Quantification revealed a dramatic decrease in the prepupal mitochondrial cross-sectional area (CSA; midgut and salivary gland and a significant increase in the number of mitochondria per cell. Moreover, ecdysone-induced mitochondrial fragmentation is mimicked ex vivo on third-instar larval wing discs by using 1 mM ecdysone for 2 hr. In addition, overexpression of Hid resulted in mitochondrial fragmentation in acridine orange-positive eye disc cells. Thus, mitochondria in Drosophila tissues fragment during PCD, as has been reported in C. elegans and mammalian cells (Goyal, 2007).
To assess the role of mitochondrial remodeling in PCD, mitochondrial morphology was temporally characterized in etoposide-, actinomycin-D-, cycloheximide-, or C6-ceramide (a lipid cell death mediator)-treated larval hemocytes and the S2R+ cell line. A 3- to 4-fold increase in nuclear condensation (6 hr) was preceded by effector caspase activation (5 hr) and phosphatidylserine (PS) exposure in propidium iodide (PI)-negative hemocytes (6 hr). These cells subsequently (10 hr) became characteristically blebbed and PI permeable. The number of etoposide-treated apoptotic hemocytes increased with time. Interestingly, mitochondrial fragmentation, as confirmed by quantifying functionally isolated mitochondria at 3 hr, preceded the onset of PS exposure or nuclear damage. Quantification showed an increase in the number of mitochondria and the contribution of fragmented mitochondria to the mitochondrial cross-sectional area (CSA). Mitochondrial fragmentation was also observed in cycloheximide- or actinomycin-D-treated, Mito-YFP-transfected S2R+ cells (Goyal, 2007).
Surprisingly, mitochondria in C6-ceramide-treated (30-60 min) hemocytes that had normal nuclei were highly contiguous. Quantifying functionally isolated mitochondrial CSA per cell showed a significant increase in the contribution of tubular or extensively tubular mitochondria in these cells when compared with untreated cells. However, by 4 hr, these extensively tubular mitochondria underwent fragmentation in FITC-Annexin V (AnV)-negative hemocytes that had normal nuclei, similar to what was observed with genotoxic stress (Goyal, 2007).
Therefore, genotoxic stresses trigger mitochondrial fragmentation, while the lipid cell death mediator induces increased mitochondrial contiguity and subsequent fragmentation prior to phosphatidylserine exposure, nuclear condensation, and finally plasma membrane permeability during Drosophila cell death (Goyal, 2007).
In hemocytes incubated with an apoptotic stimulus, mitochondrial fragmentation (3-4 hr) preceded any detectable effector caspase activation. Furthermore, inhibiting caspases with zVAD-fmk or by overexpressing DIAP-1 (DIAP-1+) did not affect mitochondrial fragmentation, although hemocyte death was inhibited, as revealed by a lack of apoptotic markers. In addition, overexpression of Dcp-1, a Drosophila effector caspase, did not affect mitochondrial morphology. Thus, mitochondrial fragmentation is upstream of effector caspase activation (Goyal, 2007).
The drp-1 mutants used to study the role of mitochondrial remodeling during Drosophila PCD are functional null alleles, drp-12 (Gly293Ser mutation), picked in a forward screen for genes affecting neurotransmission and drp-1[KG 03815], a P element insertion between the first two exons of drp-1 (13510 in this study) and a hypomorph, nrdD46 (Arg278Trp mutation; 3665 in this study). drp-12, 13510, and the deficiency Df Exel6008 were second-instar larval lethal; however, drp-12 yielded bang-sensitive escapers. The hypomorphic trans-allelic combination of 3665/13510 was third-instar larval lethal, although it yielded a few temperature-sensitive adults. A genomic duplication of drp-1 (Dp [2;1] JS13) completely rescued the lethality associated with drp-12, 13510, and 3665/13510 (Goyal, 2007).
Mitochondria in drp-12 and 3665/13510 hemocytes were extensively tubular when compared with wild-type mitochondria. Quantifying mitochondrial morphology revealed a 2-fold decrease in the number of mitochondria and a significant increase in the contribution of tubular and extensively tubular mitochondria to the total mitochondrial CSA in drp-1 mutant hemocytes when compared with wild-type cells. Interestingly, 13510/+ hemocytes or eye disc cells displayed a dominant mitochondrial fission defect that was completely rescued by a genomic duplication of drp-1. The mitochondrial fission defect in mutant cells could result from a reduced mitochondrial association of Drp-1 (Goyal, 2007).
An increase in mitochondrial contiguity due to a loss of Drp-1 function was also confirmed by measuring fluorescence recovery after photobleaching (FRAP) of Mito-YFP in drp-1 RNAi-treated S2R+ cells that had extensively tubular mitochondria. Relative FRAP of Mito-YFP in a defined mitochondrial region in drp-1 RNAi-treated cells was significantly higher than that observed in mock RNAi-treated cells (Goyal, 2007).
drp-1 mutant hemocytes were protected from etoposide-induced death up to at least 10 hr, as revealed by a lack of caspase activation, PS exposure, or PI permeability in the majority (~80%) of these cells. Furthermore, drp-1 mutant and dsRNA-treated S2R+ cells were significantly protected from cycloheximide-, actinomycin-D-, or UV-B-induced death. Consistent with increased protection, mitochondria in the majority (~98%) of etoposide-treated drp-12 hemocytes failed to fragment. Interestingly, mitochondria in etoposide-treated 3665/13510 hemocytes revealed a tubular, yet beaded and swollen intermediate in mitochondrial fragmentation by 4 hr that yielded some fragmented mitochondria in few (~25%) cells later. Therefore, reduced (drp-12) or delayed (3665/13510) mitochondrial fragmentation decreased effector caspase activation and protected cells from genotoxic stress. Moreover, an increase in expression of Drp-1 in hemocytes resulted in enhancement of etoposide-induced cell death (Goyal, 2007).
The majority (~70%) of the C6-ceramide-treated drp-12 hemocytes did not show effector caspase activation or PS exposure and displayed significant protection, similar to what was observed with etoposide, although hemocytes derived from the weaker allelic combination, 13510/3665, were apoptotic. Unlike 13510/3665 mitochondria, drp-12 mitochondria failed to fragment, consistent with an essential role for Drp-1-mediated mitochondrial fragmentation during apoptosis in Drosophila. Moreover, developmental PCD in drp-12 mutant larvae was considerably reduced, as revealed by the enlarged central nervous system and a prominently elongated ventral ganglion, similar to other PCD-defective mutants reported (Goyal, 2007).
During metamorphosis, the first ecdysone pulse triggers mitochondrial fragmentation in prepupal tissues, although it is after the second ecdysone pulse that salivary gland histolysis occurs. It is likely that DIAP-1 inhibits caspases in these cells that have fragmented mitochondria until it is downregulated at the transcriptional level or degraded after the second ecdysone pulse. Interestingly, this was mimicked ex vivo in etoposide-treated DIAP-1+ hemocytes (Goyal, 2007).
The data presented in this study show involvement of mitochondrial fragmentation for ARK-mediated Dronc activation during cell death. The RHG-family proteins that localize to the mitochondria might activate Drp-1-mediated mitochondrial fragmentation. This could result in exposure of cytochrome-C or release of Peanut, which antagonize DIAP-1-mediated suppression of Dronc. However, since Drosophila PCD is unaffected upon knockdown of cytochrome-C, mitochondrial fragmentation in Drosophila and mammalian cells would increase mitochondrial surface area and perhaps the concentration of bulky head group lipids on the outer mitochondrial membrane, facilitating recruitment of proapoptotic proteins. Drp-1 might organize sites for Drosophila Bcl-2-family protein Debcl function on mitochondria that are similar to mitochondrial sites of Bax recruitment in mammalian cells (Goyal, 2007 and references therein).
These results provide the first evidence that Drp-1-mediated mitochondrial fragmentation upstream of effector caspase activation modifies apoptotic sensitivity. Thus, mitochondrial fragmentation, like caspase activation, plays a conserved and unifying role in diverse cell death pathways from worms to mammals. Although the function of the highly contiguous mitochondria during lipid-induced cell death remains poorly understood, this study brings to the forefront a modulatory role for mitochondrial remodeling in determining the susceptibility of Drosophila cells to death.
Mitochondrial disruption is a conserved aspect of apoptosis, seen in many species from mammals to nematodes. Despite significant conservation of other elements of the apoptotic pathway in Drosophila, a broad role for mitochondrial changes in apoptosis in flies remains unconfirmed. This study shows that Drosophila mitochondria become permeable in response to the expression of Reaper and Hid, endogenous regulators of developmental apoptosis. Caspase activation in the absence of Reaper and Hid is not sufficient to permeabilize mitochondria, but caspases play a role in Reaper- and Hid-induced mitochondrial changes. Reaper and Hid rapidly localize to mitochondria, resulting in changes in mitochondrial ultrastructure. The dynamin-related protein, Dynamin related protein 1 (Drp1), is important for Reaper- and DNA-damage-induced mitochondrial disruption. Significantly, it was shows that inhibition of Reaper or Hid mitochondrial localization or inhibition of Drp1 significantly inhibits apoptosis, indicating a role for mitochondrial disruption in fly apoptosis (Abdelwahid, 2007).
A role for mitochondria in apoptosis appears to be conserved from mammals to nematodes to yeast. The lack of clear evidence that mitochondria play a role in Drosophila apoptosis has prompted discussion of whether flies represent an evolutionary outlier in this highly conserved process. The data strongly suggests that mitochondrial disruption also plays a role in Drosophila apoptosis (Abdelwahid, 2007).
The data show that mitochondria rapidly become permeable to Cyt c when Rpr or Hid are expressed, both in cultured cells and in vivo. This alteration in mitochondrial permeability was also seen during DNA-damage-induced apoptosis. Importantly, it was demonstrated that the mitochondrial permeabilization during DNA-damage-induced apoptosis is dependent on the genes in the H99 interval. Taken together, these data indicate that Rpr and Hid are both necessary and sufficient for mitochondrial permeabilization (Abdelwahid, 2007).
In contrast, apoptosis induced by Actinomycin D, UV, and DIAP1 RNAi does not result in mitochondrial permeabilization. This indicates that caspase activation alone is not sufficient to induce mitochondrial permeabilization and that the mitochondrial permeabilization seen on Rpr or Hid induction is not simply a general late event in apoptosis. The efficient cell killing by Actinomycin D, UV, and DIAP1 RNAi also implies that mitochondrial permeabilization is not important for all apoptosis in Drosophila cells. Rather, it suggests that the Rpr and Hid proteins have a specific activity on the mitochondria that results in mitochondrial permeabilization to execute apoptosis in a timely manner (Abdelwahid, 2007).
The effects of Rpr and Hid on mitochondria were not limited to permeabilization. It was found that mitochondrial morphology is dramatically altered within 90 min of Rpr or Hid expression, in both S2 cells and embryos. A variety of defects were found in mitochondrial ultrastructure ranging from a rounded appearance, to bulging (and occasional rupture) of the outer mitochondrial membrane, to swelling of the matrix and disruption of the cristae. This was rarely seen with other inducers of apoptosis. Rpr and Hid may directly cause altered mitochondrial morphology or could act indirectly through other proteins localized at the mitochondria (Abdelwahid, 2007).
The absence of mitochondrial permeabilization in cells treated with DIAP1 dsRNA indicates that the mitochondrial function of Rpr and Hid is independent of their ability to inhibit DIAP1. This is confirmed by data showing that expression of DeltaN-Rpr results in mitochondrial permeabilization despite the fact that this protein lacks the necessary motif to inhibit DIAP1 antiapoptotic activity. Taken together, these data demonstrate that Rpr and Hid have dual activities in the cell, both to inhibit DIAP1 and to permeabilize mitochondria. Data from other labs have suggested that Rpr is a multifunctional protein. The data confirm that Rpr has multiple proapoptotic activities in the fly (Abdelwahid, 2007).
The dual functionality of Rpr and Hid parallel the recently described role of C. elegans Egl-1 in mitochondrial damage. Egl-1 induces apoptosis by binding to Ced-9 to promote both the activation of the caspase Ced-3 and mitochondrial fragmentation. Similarly, Rpr and Hid bind to DIAP1, displacing active caspases and act on mitochondria to promote mitochondrial disruption. One difference between C. elegans and flies appears to be the requirement for caspase activity in the mitochondrial disruption. In C. elegans, Ced-3 is not required for fragmentation but is required for apoptosis in response to fragmentation. In Drosophila, caspase activity participates in the mitochondrial changes (Abdelwahid, 2007).
Two lines of evidence support a role for mitochondrial disruption in Drosophila apoptosis. First, Rpr and Hid must localize to mitochondria to elicit a full apoptotic response. Second, if mitochondrial disruption is blocked by inhibiting Dynamin related protein 1 (Drp1) expression, a decrease is seen in apoptosis. These data clearly indicate that mitochondrial localization of Rpr and Hid is required for a full apoptotic response in S2 cells. This agrees with previous data on Rpr and also with studies on a Grim mutant lacking a mitochondrial localization signal. Mitochondrial localization of Hid has been demonstrated in a heterologous system. In the Haining study, Hid killing was not compromised in the absence of mitochondrial localization, in contrast to the current observations in Drosophila cells. A role for mitochondrial localization is also supported by the finding that two mutant forms of Hid that lack mitochondrial localization in mammalian cells behave as weak loss-of-function alleles in the fly (Abdelwahid, 2007).
The mitochondrial fission protein Drp1 is implicated in mitochondrial disruption during apoptosis in yeast, nematodes, and mammals. The current data indicate a role for this protein in Rpr-induced and DNA-damage-induced mitochondrial disruption in S2 cells and in the embryo. Furthermore, the inhibition of mitochondrial disruption after Drp1 knockdown is correlated with a decrease in apoptosis, strongly suggesting that mitochondrial disruption contributes to the apoptotic response. It is interesting to note that Drp1 plays a conserved role in apoptosis in a wide variety of organisms but seems to function downstream of different pathways. In mammals, inhibition of Drp1 blocks apoptosis in response to activation of proapoptotic Bcl-2 family members. In C. elegans, Drp1 inhibition blocks endogenous death downstream of Egl1 and Ced9, also Bcl-2 family proteins. Even in yeast, the role of Drp1 in cell death can be modulated by Bcl-2 family proteins. Surprisingly, in flies, Drp1 appears to be acting downstream of a different family of apoptosis inducers, the RHG proteins. It remains to be seen whether a role for the fly Bcl-2 family proteins can be established in mitochondrial disruption (Abdelwahid, 2007).
Release of apoptogenic factors, most notably Cyt c, from the mitochondria is an essential step in most apoptosis in mammalian systems. However, the current work confirms the findings of others that Cyt c, although released from mitochondria by Rpr and Hid, is not important for Rpr or Hid killing. It should be noted that Cyt c has been shown to be important in some Drosophila developmental apoptosis. In these deaths, Hid is likely to act upstream of Cyt c release. If Cyt c release is required in some cells for Hid-mediated caspase activation, why not in S2 cells? It is possible that there are both Cyt c-dependent and -independent mechanisms for activating caspases, and these may be cell-type dependent. Recent data from mice carrying a nonapoptogenic form of Cyt c supports this model, since this study suggests that there is both Cyt c-dependent and -independent apoptosis during mouse development (Abdelwahid, 2007).
If release of Cyt c is not an essential step in apoptosis in most fly cells, is another apoptosis-inducing factor released during mitochondrial disruption? In mammalian cells, release of other mitochondrial proteins such as SMAC/Diablo, Omi/HTRA2, and AIF are proposed to contribute to apoptosis. There is some evidence that released mitochondrial factors do not contribute to caspase activation in the fly. Unlike in the mammalian system, mitochondrial lysates cannot activate caspases in fly cytoplasmic lysates. An alternative possibility is that mitochondrial disruption per se might contribute to apoptosis in the fly through inhibition of normal mitochondrial functions essential for cell viability. This might serve as a backup system, to maximize apoptosis in cells that express low levels of the RHG proteins. A similar role for mitochondrial disruption has been proposed in C. elegans (Abdelwahid, 2007).
In sum, it is concluded from these studies that Drosophila is not an outlier in evolution with regard to the involvement of mitochondria in the apoptotic process. Rather, the data indicate that mitochondrial changes contribute to Drosophila apoptosis. The findings suggest that the view of the role of mitochondria in cell death has to be broadened beyond the release of proapoptotic factors, to include the general disruption of mitochondria, ensuring that doomed cells have no chance of recovery. Such a model would fit not only the changes seen in mammalian mitochondria, but also those found in yeast, C. elegans, and flies as well (Abdelwahid, 2007).
During the cell cycle, mitochondria undergo regulated changes in morphology. Two particularly interesting events are first, mitochondrial hyperfusion during the G1-S transition and second, fragmentation during entry into mitosis. The mitochondria remain fragmented between late G2- and mitotic exit. This mitotic mitochondrial fragmentation constitutes a checkpoint in some cell types, of which little is known. This study bypassed the 'mitotic mitochondrial fragmentation' checkpoint by inducing fragmented mitochondrial morphology and then measuring the effect on cell cycle progression. Using Drosophila larval hemocytes, Drosophila S2R+ cell and cells in the pouch region of wing imaginal disc of Drosophila larvae it was shown that inhibiting mitochondrial fusion, thereby increasing fragmentation, causes cellular hyperproliferation and an increase in mitotic index. However, mitochondrial fragmentation due to over-expression of the mitochondrial fission machinery does not cause these changes. These experiments suggest that the inhibition of mitochondrial fusion increases superoxide radical content and leads to the upregulation of cyclin B that culminates in the observed changes in the cell cycle. Evidence is provided for the importance of mitochondrial superoxide in this process. These results provide an insight into the need for mitofusin-degradation during mitosis and also help in understanding the mechanism by which mitofusins may function as tumor suppressors (Gupte, 2015).
Changes in mitochondrial dynamics (fusion and fission) are known to occur during stem cell differentiation; however, the role of this phenomenon in tissue aging remains unclear. This study reports that mitochondrial dynamics are shifted toward fission during aging of Drosophila ovarian germline stem cells (GSCs), and this shift contributes to aging-related GSC loss. As GSCs age, mitochondrial fragmentation and expression of the mitochondrial fission regulator, Dynamin-related protein (Drp1), are both increased, while mitochondrial membrane potential is reduced. Moreover, preventing mitochondrial fusion in GSCs results in highly fragmented depolarized mitochondria, decreased BMP stemness signaling, impaired fatty acid metabolism, and GSC loss. Conversely, forcing mitochondrial elongation promotes GSC attachment to the niche. Importantly, maintenance of aging GSCs can be enhanced by suppressing Drp1 expression to prevent mitochondrial fission or treating with rapamycin, which is known to promote autophagy via TOR inhibition. Overall, these results show that mitochondrial dynamics are altered during physiological aging, affecting stem cell homeostasis via coordinated changes in stemness signaling, niche contact, and cellular metabolism. Such effects may also be highly relevant to other stem cell types and aging-induced tissue degeneration (Amartuvshin, 2020).
It has become evident that caspases function in nonapoptotic cellular processes in addition to the canonical role for caspases in apoptotic cell death. It has been demonstrated that the Drosophila effector caspase Dcp-1 localizes to the mitochondria and positively regulates starvation-induced autophagic flux during mid-oogenesis. Loss of Dcp-1 leads to elongation of the mitochondrial network, increased levels of the adenine nucleotide translocase sesB, increased ATP levels, and a reduction in autophagy. sesB is a negative regulator of autophagic flux, and Dcp-1 interacts with sesB in a nonproteolytic manner to regulate its stability, uncovering a novel mechanism of mitochondrial associated, caspase-mediated regulation of autophagy in vivo (DeVorkin, 2014).
Mitochondrial AAA (ATPases Associated with diverse cellular Activities) proteases i-AAA (intermembrane space-AAA) and m-AAA (matrix-AAA) are closely related and have major roles in inner membrane protein homeostasis. Mutations of m-AAA proteases are associated with neuromuscular disorders in humans. However, the role of i-AAA in metazoans is poorly understood. This study generated a deletion affecting Drosophila i-AAA, dYME1L (dYME1Ldel). Mutant flies exhibited premature aging, progressive locomotor deficiency and neurodegeneration that resemble some key features of m-AAA diseases. dYME1Ldel flies displayed elevated mitochondrial unfolded protein stress and irregular cristae. Aged dYME1Ldel flies had reduced complex I (NADH/ubiquinone oxidoreductase) activity, increased level of reactive oxygen species (ROS), severely disorganized mitochondrial membranes and increased apoptosis. Furthermore, inhibiting apoptosis by targeting dOmi (Drosophila Htra2/Omi) or DIAP1, or reducing ROS accumulation suppressed retinal degeneration. The results suggest that i-AAA is essential for removing unfolded proteins and maintaining mitochondrial membrane architecture. Loss of i-AAA leads to the accumulation of oxidative damage and progressive deterioration of membrane integrity, which might contribute to apoptosis upon the release of proapoptotic molecules such as dOmi. Containing ROS level could be a potential strategy to manage mitochondrial AAA protease deficiency (Qi, 2015).
E2F/DP transcription factors regulate cell proliferation and apoptosis. This study investigated the mechanism of the resistance of Drosophila dDP mutants to irradiation-induced apoptosis. Contrary to the prevailing view, this is not due to an inability to induce the apoptotic transcriptional program, because this program is induced; rather, this is due to a mitochondrial dysfunction of dDP mutants. This defect is attributed to E2F/DP-dependent control of expression of mitochondria-associated genes. Genetic attenuation of several of these E2F/DP targets mimics the dDP mutant mitochondrial phenotype and protects against irradiation-induced apoptosis. Significantly, the role of E2F/DP in the regulation of mitochondrial function is conserved between flies and humans. Thus, these results uncover a role of E2F/DP in the regulation of mitochondrial function and demonstrate that this aspect of E2F regulation is critical for the normal induction of apoptosis in response to irradiation (Ambrus, 2013).
E2F transcription factors are best understood for their role in controlling the cell cycle, apoptosis, and differentiation. In this report, evidence is presented that E2F is also involved in the regulation of mitochondrial function and identify a specific biological context, DNA damage-induced apoptosis, in which this aspect of E2F control becomes critical. It is suggested that mitochondrial dysfunction, and not the failure to induce the apoptotic geneexpression program, makes E2F-deficient cells refractory to apoptosis (Ambrus, 2013).
In flies and mammals, the conserved mechanism by which E2F triggers apoptosis is transcriptional control of apoptotic targets. Therefore, it is believed that in irradiated cells, dE2f1, like dp53, contributes to the normal transcriptional induction of apoptotic genes. However, the current data do not support such a model since the apoptotic gene expression program was induced properly in irradiated dDP mutants. Thus, in the context of the DNA damage response, the contribution of dE2f1 to the normal transcriptional induction of apoptotic genes is negligible. It is emphasized that the data do not imply that dE2f1 is unimportant.
For example, unrestrained dE2f1 activity in rbf mutants has been shown to markedly increase the induction of hid and rpr in response to DNA damage, and this increase determines the elevated sensitivity of rbf mutants to irradiation-induced apoptosis. Since ablation of dp53 completely blocks irradiation-induced apoptosis and induction of apoptotic genes, the irradiation-induced apoptotic program is governed primarily by dp53, and hyperactive dE2f1 can provide additional assistance to dp53 in activating apoptotic genes. This contribution of dE2f1 becomes evident in certain settings, such as in rbf mutants (Ambrus, 2013).
Given that irradiated dDP mutants have a properly induced apoptotic gene expression program that should trigger apoptosis in these cells, their failure to undergo apoptosis is puzzling. It is suggested that the resistance of dDP mutants to apoptosis is the consequence of a mitochondrial dysfunction. In dDP mutants, mitochondria exhibit an abnormal morphology and reduced mitochondrial membrane potential and ATP levels. The lower level of expression of dE2f/dDP mitochondria-associated target genes is a critical event in determining the response of dDP mutants to irradiation, since genetic attenuation of their expression mimics the dDP mutant mitochondrial phenotype and protection from irradiation-induced apoptosis. Significantly, the strongest protection was observed with the genes that exerted the most severe mitochondrial defects upon downregulation. Thus, the response to irradiation-induced apoptosis correlates with the extent of the mitochondrial defects. One possibility is that the mitochondrial dysfunction of dDP-deficient cells lowers their mitochondrial readiness for apoptosis, and therefore the irradiation-induced apoptotic transcriptional program is insufficient to trigger cell death. Intriguingly, 'mitochondrial readiness for apoptosis' is thought to be the molecular basis of a differential response to chemotherapy in cancer patients with acute myelogenous leukemia. Among the mitochondria-associated dE2F/dDP target genes investigated in this work, Mdh2 is particularly interesting. It was previously shown that an Mdh2 mutation prevented apoptosis in another context during ecdysone-induced cell death in salivary glands. Destruction of salivary glands is normally triggered by the induction of rpr and rpr, and both genes were induced in Mdh2 mutants to the level observed in the wild-type. In addition, Mdh2 mutants display a defect in energy production and reduced ATP levels, which is thought to compromise their ability to undergo apoptosis. This setting is highly reminiscent of dDP mutants, which are also remarkably resistant to cell death even in the face of a high level of induction of a DNA damage-dependent apoptotic transcription program (Ambrus, 2013).
The idea that mitochondrial defects could impact execution of apoptosis is consistent with the recently uncovered importance of mitochondria for cell death in Drosophila. Several studies demonstrated that Rpr, Grim, and Hid, the key apoptotic proteins in flies, are localized to mitochondria and that this localization is required for efficient activation of apoptosis. Thus, one possibility is that proapoptotic proteins are not efficiently localized to mitochondria in dDP mutants. It is also possible that the dysfunctional mitochondria of dDP mutants fail to remodel in response to irradiation, which has been shown to be necessary for execution of stress-induced apoptosis. It is noted that the current findings do not imply that dE2F/ dDP normally triggers apoptosis by modulating mitochondrial function, but rather that mitochondrial function is compromised in E2F-deficient cells, which in turn would result in less efficient apoptosis (Ambrus, 2013).
Another important conclusion of this study is that a mechanistic link between the Rb pathway and mitochondria is conserved in mammalian cells. Several Drosophila dE2F/dDP-regulated, mitochondria-associated genes are also E2F targets in mammalian cells, and their expression is similarly reduced in cells when E2F is inactivated. Significantly, this leads to strong mitochondrial defects that are highly reminiscent of the mitochondrial phenotype in Drosophila dDP mutant eye discs. The data are consistent with the recent finding that mammalian E2f1 and pRB regulate expression of oxidative metabolism genes during the adaptive metabolic response in mice. The Rb pathway has been also implicated in the regulation of the mitochondrial biogenesis transcriptional program in erythropoiesis. Intriguingly, a recent study demonstrated that a fraction of endogenous pRB is present at mitochondria, where it directly participates in mitochondrial apoptosis (Hilgendorf, 2013). Given the prominent role of mitochondrial pathways in apoptosis in mammalian cells, it is conceivable that the loss of E2F could impact the efficacy of apoptosis in mammals by a mechanism analogous to that observed in Drosophila. Such an idea is consistent with the finding that inactivation of E2F reduces DNA damage-induced apoptosis in mammalian cells (Ambrus, 2013).
Interestingly, Nicolay (2013) recently demonstrated that an rbf mutation alters cellular metabolism and an abnormal metabolism sensitizes Drosophila to various types of stress. This work, found that inactivation of E2F results in a severe mitochondrial dysfunction, which is the basis for the failure of dDP mutants to undergo DNA damage-induced apoptosis. Thus, a general emerging theme is that perturbation of the Rb pathway may exert profound metabolic changes within the cell that can have a major impact on cell survival (Ambrus, 2013).
The heart is a muscle with high energy demands. Hence, most
patients with mitochondrial disease produced by defects in the
oxidative phosphorylation (OXPHOS) system are susceptible to
cardiac involvement. The presentation of mitochondrial
cardiomyopathy includes hypertrophic, dilated and left ventricular
noncompaction, but the molecular mechanisms involved in cardiac
impairment are unknown. One of the most frequent OXPHOS defects in
humans frequently associated with cardiomyopathy is cytochrome c
oxidase (COX) deficiency caused by mutations in COX assembly
factors such as Sco1 and Sco2. To investigate the molecular
mechanisms that underlie the cardiomyopathy associated with Sco
deficiency, this study interfered with scox (the single
Drosophila Sco orthologue) expression in the heart.
Cardiac-specific knockdown of scox reduces fly lifespan, and it
severely compromises heart function and structure, producing
dilated cardiomyopathy. Cardiomyocytes with low levels of scox
have a significant reduction in COX activity and they undergo a
metabolic switch from OXPHOS to glycolysis, mimicking the clinical
features found in patients harbouring Sco mutations. The major
cardiac defects observed are produced by a significant increase in
apoptosis, which is dp53-dependent.
Genetic and molecular evidence strongly suggest that dp53 is
directly involved in the development of the cardiomyopathy induced
by scox deficiency. Remarkably, apoptosis is enhanced in the
muscle and liver of Sco2 knock-out mice, clearly suggesting that
cell death is a key feature of the COX deficiencies produced by
mutations in Sco genes in humans (Martínez-Morentin, 2015).
Cardiomyopathies are a collection of myocardial disorders in which
the heart muscle is structurally and functionally abnormal. In the
past decade, it has become clear that an important proportion of
cases of hypertrophic and dilated cardiomyopathies are caused by
mutations in genes encoding sarcomeric or desmosomal proteins. In
addition, cardiomyopathies (both hypertrophic and dilated) are
frequently associated to syndromic and non-syndromic mitochondrial
diseases. The importance of oxidative metabolism for cardiac
function is supported by the fact that 25–35% of the
myocardial volume is taken by mitochondria. The current view of
mitochondrial involvement in cardiomyopathy assumes that ETC
malfunction results in an increased ROS production, triggering a
“ROS-induced ROS release” vicious circle which in turn
perpetuates ETC dysfunction via damage in mtDNA and proteins
involved in electron transport. Under this view, accumulated
mitochondrial damage would eventually trigger apoptosis through
mitochondrial permeability transition pore (mPTP) opening other
mechanisms. Under normal circumstances, damaged mitochondria would
be eliminated through mitophagy. Excessive oxidative damage is
supposed to overcome the mitophagic pathway resulting in
apoptosis. Nevertheless, although several potential mechanisms
have been suggested, including apoptosis deregulation, oxidative
stress, disturbed calcium homeostasis or impaired iron metabolism,
the molecular basis of the pathogenesis of mitochondrial
cardiomyopathy is virtually unknown (Martínez-Morentin,
2015). Pathogenic mutations in human SCO1
and SCO2
have been reported to cause hypertrophic cardiomyopathy, among
other clinical symptoms. However, the molecular mechanisms
underlying this cardiac dysfunction have yet to be elucidated.
This study reports the first cardiac-specific animal model to
study human SCO1/2-mediated cardiomyopathy. Cardiac-specific scox
KD in Drosophila provokes a severe dilated
cardiomyopathy, as reflected by a significant increase in the
conical chamber size, due to mitochondrial dysfunction. It
presents a concomitant metabolic switch from glucose oxidation to
glycolysis and an increase in ROS levels, leading to p53-dependent
cell death. Interestingly, previous studies on patients and rat
models have shown that mitochondrial dysfunction is associated
with abnormalities in cardiac function and changes in energy
metabolism, resulting in glycolysis optimization and lactic
acidosis. Furthermore, in the Sco2KI/KO mouse model,
where no evidence of cardiomyopathy has been described, partial
loss of Sco2 function induces apoptosis in liver and skeletal
muscle. In flies scox KD causes a significant reduction
in FS and in the DI, as well as cardiac myofibril disorganization.
This degenerative process is most likely due to mitochondrial
dysfunction rather than to a developmental defect and moreover,
the dilated cardiomyopathy developed by flies resembles that
caused by mitochondrial fusion defects in flies
(Martínez-Morentin, 2015). The ETC is the major site of ROS production in cells, and aging
and many neurodegenerative diseases have been linked to
mitochondrial dysfunction that results in excessive oxidative
stress. Interestingly, there is an increase in ROS formation
associated with oxidative DNA damage in human Sco2−/−
cells. Accordingly, it was found that cardiac-specific knockdown
of scox increases oxidative stress, although it could
not be distinguished whether this increase in free radical
accumulation arises from the mitochondria or whether it comes from
non-mitochondrial sources due to a loss of cellular homeostasis,
as reported in yeast and in a neuro-specific COX-deficient
Alzheimer disease mouse model (Martínez-Morentin, 2015). Sco2 expression is known to be modulated by p53, a transcription
factor that participates in many different processes, including
cancer development, apoptosis and necrosis. p53 regulates
homeostatic cell metabolism by modulating Sco2 expression and
contributes to cardiovascular disorders. In addition, p53
activation in response to stress signals, such as increased
oxidative stress or high lactic acid production, is well
documented. Data from this study, showing that p53 is upregulated
in response to scox KD, but not in response to KD of
another Complex IV assembly factor, Surf1,
suggest a specific genetic interaction between dp53 and
scox. This is corroborated by the dramatic effects
observed in the heart structure and function when dp53 is
overexpressed in scox KD hearts. Furthermore, the
functional and structural defects seen in scox KD hearts
can be rescued in dp53-DN OE or dp53 null
backgrounds, indicating that the scox-induced defects are mediated
by increased p53 expression. Interestingly, opposed to scox
KD, the heart structure defects induced by dp53 OE can
be fully rescued by heart-specific Surf1 KD, further
confirming the specificity of the genetic interaction between dp53
and scox (Martínez-Morentin, 2015). It has recently been shown that SCO2 OE induces
p53-mediated apoptosis in tumour xenografts and cancer cells.
Furthermore, SCO2 KD sensitizes glioma cells to
hypoxia-induced apoptosis in a p53-dependent manner and induces
necrosis in tumours expressing WT p53, further linking the
SCO2/p53 axis to cell death. In Drosophila, there is a
dp53-mediated upregulation of Reaper,
Hid and Grim
in response to scox KD. This, coupled with the
observation that Reaper overexpression in the adult heart enhances
the structural defects caused by cardiac-specific scox
KD, suggests that scox normally prevents the triggering of
dp53-mediated cell death in cardiomyocytes in stress response.
Indeed, it was found that there is massive cell death in the
skeletal muscle and liver of Sco2KI/KO mice, supporting
the hypothesis that Sco proteins might play this role also in
mammals (Martínez-Morentin, 2015). The study provides evidence that scox KD hearts exhibit
partial loss of COX activity, with cardiomyocytes undergoing
apoptosis. There is evidence from vertebrate and invertebrate
models that partial inhibition of mitochondrial respiration
promotes longevity and metabolic health due to hormesis. In fact,
it has recently been shown that mild interference of the OXPHOS
system in Drosophila IFMs preserves mitochondrial
function, improves muscle performance and increases lifespan
through the activation of the mitochondrial unfolded pathway
response and IGF/like
signalling pathways. This study speculates that cell death, rather
than mitochondrial dysfunction itself, is likely to be the main
reason for the profound heart degeneration observed in TinCΔ4-Gal4>scoxi
flies. Expression of dominant negative dp53 in scox
KD hearts rescues dysfunction and cardiac degeneration, and, most
importantly, scox KD in dp53−/−
animals causes no apparent heart defects, which could attribute
the rescue observed to blockade of the p53 pathway. Indeed,
inhibiting apoptosis by p35 or Diap1
OE almost completely rescues the morphological scox KD
phenotype. As scox KD in the absence of dp53 causes no
symptoms of heart disease, coupled with the inability of p35 and
Diap1 to completely rescue the morphological phenotype, suggests
that, in addition to inducing apoptosis, dp53 plays a
key role in the development of cardiomyopathy
(Martínez-Morentin, 2015). The fact that heart-specific Surf1 KD neither
upregulates p53 nor induces apoptosis supports the idea that the
partial loss of scox function itself triggers dp53
upregulation and apoptosis, rather than it being a side effect of
COX dysfunction and the loss of cellular homeostasis. In this
context, it is noteworthy that SCO2 interference in
mammalian cells induces p53 re-localization from mitochondria to
the nucleus. It is therefore tempting to hypothesize that scox
might play another role independent of its function as a COX
assembly factor, perhaps in redox regulation as suggested
previously and that it may act in conjunction with dp53 to fulfil
this role. Another issue deserves further attention, the
possibility of this interaction being a tissue-specific response.
It may be possible that the threshold of COX deficiency tolerated
by the heart might be lower than in other tissues, thus the scox/dp53
genetic interaction may be a tissue-dependent phenomenon or the
consequence of a tissue-specific role of scox. In fact,
mitochondrial dysfunction in mice is sensed independently from
respiratory chain deficiency, leading to tissue-specific
activation of cellular stress responses. Thus, more work is
necessary to test these hypotheses and try to understand how the
partial lack of scox induces cell death through dp53
(Martínez-Morentin, 2015). Although the role of mitochondria in Drosophila
apoptosis remains unclear, there is strong evidence that, as in
mammals, mitochondria play an important role in cell death in
flies. The localization of Rpr, Hid and Grim in the mitochondria
is essential to promote cell death, and fly mitochondria undergo
Rpr-, Hid- and Drp1-dependent
morphological changes and disruption following apoptotic stimulus.
Moreover, the participation of the mitochondrial fission protein
Drp1 in cell death is conserved in worms and mammals. It has been
proposed that p53 plays a role in the opening of the mPTP that
induces necrotic cell death. According to this model, p53
translocates to the mitochondrial matrix upon ROS stimulation,
where it binds cyclophilin D (CypD) to induce mPTP opening
independent of proapoptotic Bcl-2 family members Bax and Bak, and
in contrast to traditional concepts, independent of Ca2+
(Martínez-Morentin, 2015). Apoptotic and necrotic pathways have a number of common steps and
regulatory factors, including mPTP opening that is thought to
provoke mitochondrial swelling and posterior delivery of necrotic
factors, although Drosophila mPTP activation is not
accompanied by mitochondrial swelling. Interestingly, although the
p53 protein triggers mitochondrial outer membrane permeabilization
(MOMP) in response to cellular stress in mammals, releasing
mitochondrial death factors, MOMP in Drosophila is more
likely a consequence rather than cause of caspase activation and
the release of mitochondrial factors does not appear to play a
role in apoptosis. Thus, in cardiac-specific scox KD
flies, dp53 might induce mPTP opening to trigger cell death, which
in the absence of mitochondrial swelling would result in apoptosis
instead of necrosis, as occurs in mammals. Drosophila
mPTP has been shown to be cyclosporine A (CsA)-insensitive in
vitro, although CsA administration ameliorates the mitochondrial
dysfunction with a severely attenuated ATP and enhanced ROS
production displayed by collagen XV/XVIII mutants. Interestingly,
mice lacking collagen VI display altered mitochondrial structure
and spontaneous apoptosis, defects that are caused by mPTP opening
and that are normalized in vivo by CsA treatment
(Martínez-Morentin, 2015). Hypoplastic left heart syndrome (HLHS) is a severe congenital heart disease (CHD) with a likely oligogenic etiology, but understanding of the genetic complexities and pathogenic mechanisms leading to HLHS is limited. This study performed whole genome sequencing (WGS) on 183 HLHS patient-parent trios to identify candidate genes, which were functionally tested in the Drosophila heart model. Bioinformatic analysis of WGS data from an index family of a HLHS proband born to consanguineous parents prioritized 9 candidate genes with rare, predicted damaging homozygous variants. Of them, cardiac-specific knockdown (KD) of mitochondrial MICOS complex subunit dCHCHD3/6 resulted in drastically compromised heart contractility, diminished levels of sarcomeric actin and myosin, reduced cardiac ATP levels, and mitochondrial fission-fusion defects. These defects were similar to those inflicted by cardiac KD of ATP synthase subunits of the electron transport chain (ETC), consistent with the MICOS complex's role in maintaining cristae morphology and ETC assembly. Five additional HLHS probands harbored rare, predicted damaging variants in CHCHD3 or CHCHD6. Hypothesizing an oligogenic basis for HLHS, 60 additional prioritized candidate genes from these patients were tested for genetic interactions with CHCHD3/6 in sensitized fly hearts. Moderate KD of CHCHD3/6 in combination with Cdk12 (activator of RNA polymerase II), RNF149 (goliath, E3 ubiquitin ligase), or SPTBN1 (β-Spectrin, scaffolding protein) caused synergistic heart defects, suggesting the likely involvement of diverse pathways in HLHS. Further elucidation of novel candidate genes and genetic interactions of potentially disease-contributing pathways is expected to lead to a better understanding of HLHS and other CHDs (Birker, 2023).
Hypoplastic left heart syndrome (HLHS) is a birth defect that accounts for 2-4% of congenital heart defects (CHDs), equal to 1000-2000 HLHS births in the United States per year. HLHS has been proposed to be caused by genetic, epigenetic, or environmental factors. The severe cardiac characteristics of HLHS include aortic and mitral stenosis or atresia, and reduced size of the left ventricle and aorta; however, there is a spectrum of cardiac phenotypes that can underly HLHS pathophysiology. If not treated with reconstructive heart surgeries or cardiac transplantation, infants born with HLHS will not survive. To date, the standard treatment for this disease is a three-stage surgical procedure, which begins neonatally and aims overall to achieve right ventricle-dependent systemic circulation and deliver oxygen-poor blood more directly to the lungs. Although the surgical procedures correctly divert left ventricular function to the right ventricle, there is a subgroup of HLHS patients who are at risk of latent heart failure, which is often preceded by reduced ejection fraction (Birker, 2023).
Although several studies have examined the molecular underpinnings of HLHS, the number of genes associated with this disease is small (e.g. NKX2-5, NOTCH1, ETS1, MYH6, LRP2, and CELSR1), and they are not yet conclusively determined as causal for HLHS. Defining pathogenic mechanisms has proved elusive given the oligogenic complexity of HLHS. Overall, there is a great need to functionally evaluate newly emerging HLHS candidate genes to understand how they may contribute to the molecular, cellular, and morphological processes underlying HLHS (Birker, 2023).
Drosophila is well-suited for modeling genetic underpinnings of CHDs: many of the genes and gene programs found in the Drosophila heart are evolutionarily conserved, including a core set of cardiogenic transcription factors and inductive factors (e.g. Nkx2-5/tinman), approximately 75% of known human disease-causing genes having fly orthologs, and the developing mammalian and Drosophila hearts share developmental similarities, such as their origin within the mesoderm (Birker, 2023).
Mitochondria have been postulated to play a critical role in HLHS pathogenesis. For example, a recent study reported that cardiomyocytes derived from iPSCs of HLHS patients (iPSC-CM), who later developed right ventricular failure, had reduced mitochondrial concentration, ATP production, and contractile force. This study revealed downregulated expression of genes involved in mitochondrial processes, such as ATP synthesis coupled electron transport. Another study of HLHS patient-derived iPSC-CMs revealed reduced mitochondrial size, number, and malformed mitochondrial inner membranes using transmission electron microscopy. Similarly, an HLHS mouse model with Sap130 and Pcdha9 mutations showed mitochondrial defects manifested as reduced cristae density and smaller mitochondrial size. Despite a lack of understanding of the exact mitochondrial mechanisms underlying HLHS pathogenesis, recent experimental and bioinformatic data suggest an underlying role of mitochondria in HLHS (Birker, 2023).
In this study, a cohort of 183 HLHS proband-parent trios underwent whole genome sequencing (WGS) to identify candidate genes, including a prioritized consanguineous family where genes harboring rare, predicted damaging homozygous variants were investigated. Among the resulting candidate HLHS genes tested in Drosophila, cardiac-specific knockdown (KD) of Chchd3/6 (coiled-coil-helix-coiled-coil-helix-domain-containing protein 6) of the MICOS (mitochondrial contact site and cristae organization system) complex exhibited severe heart structure and function defects. The MICOS complex is an eight-subunit complex in mammals (five in Drosophila) located in the inner mitochondrial membrane that is necessary to maintain cristae morphology and ATP production. It is closely associated and interacts with SAMM50 (sorting and assembly machinery, CG7639), which is located in the outer mitochondrial membrane. The MICOS complex's role in cardiac development and functional homeostasis is not known but is likely important for efficient ATP production. Reduced contractility was observed upon cardiac-specific Chchd3/6 KD, diminished sarcomeric Actin and Myosin levels, as well as severe mitochondrial morphology defects, which manifested as fragmented and aggregated structures. Similar phenotypes were observed upon cardiac KD of other MICOS complex genes, as well as other mitochondrial genes such as ATP synthase (complex V), specifically ATP synthase B and β. Significantly diminished proliferation of human induced pluripotent stem cell (iPSC)-derived ventricular-like cardiomyocytes (VCMs) was found upon KD of MICOS genes. Finally, a family-based candidate gene interaction screen in Drosophila revealed three genes that genetically interact with Chchd3/6: Cdk12 (activator RNA polymerase II activator), RNF149 (goliath, gol, E3 ubiquitin ligase), SPTBN1 (β Spectrin, scaffolding protein). In summary, Chchd3/6 and other components important for mitochondrial homeostasis were identified as critical for establishing and maintaining cardiac structure and function, and likely contribute to HLHS and/or latent heart failure following surgical palliation (Birker, 2023).
HLHS is characterized by a small left heart, including reduced left ventricle size and mitral and/or atrial atresia or stenosis, and aortic hypoplasia, collectively obstructing systemic blood flow. As a consequence, newborns cannot sustain systemic blood flow for more than a few days and therefore require treatment soon after birth. There is a need for improved therapies to treat HLHS patients, and this requires a better understanding of the biology behind HLHS pathogenesis. This study probed the genetic basis of HLHS using WGS and powerful bioinformatic gene variant prioritization in a large cohort of HLHS proband-parent trios combined with model system validation (Birker, 2023).
The 11 H family was prioritized because of consanguinity, implicating a homozygous recessive mode of inheritance that resulted in a short list of nine candidate genes. These candidate genes were probed in Drosophila and iPSC-CMs for a potential role in cardiomyocyte development and function, to gain new insights into HLHS and CHDs in general. Among these HLHS gene candidates, this study focused on CHCHD3/6, which has not been previously studied in the heart, and which had striking cardiac functional and structural defects in Drosophila. Specifically, the preliminary gene screen demonstrated that Chchd3/6 cardiac-specific KD caused reduced contractility and decreased sarcomeric F-Actin and Myosin staining (Birker, 2023).
The data suggest that Chchd3/6 is necessary during larval and early adult stages to maintain contractility in the adult heart. This is relevant since patients with HLHS have both structural heart disease and risk for later myocardial failure. The prevailing 'no flow, no grow' hypothesis for HLHS pathogenesis surmises that reduced blood flow in the fetal heart causes underdevelopment of the left ventricle. A reduced ability for the heart to contract in utero, due to reduced CHCHD6 activity, could contribute to decreased ventricular blood flow in the embryo, resulting in an abnormally small left ventricle. Moreover, reduced CHCHD6 activity could compromise right ventricular function later in life. In fact, the 11 H proband exhibited mildly reduced right ventricular ejection fraction several years after successful surgical palliation. Consistent with the model system, CHCHD6 deficiency could result in cumulative impairment of mitochondrial function, leading to contractile dysfunction. Why a mitochondrial defect would have a preferential effect on the left ventricle is still an enigma. It is speculated that some of the patient-specific variants that potentially contribute to this likely polygenic disease are in genes that may have a higher expression level or functional importance in the left ventricle, thus in combination with MICOS variants preferentially affecting left-ventricular growth and differentiation, leading to decreased contractility, then again compounded by impaired blood flow feeding back to diminishing growth. Future studies investigating the polygenic basis of HLHS are needed to address this question (Birker, 2023).
Chchd3/6 KD in the fly heart led to mitochondrial fission-fusion defects, with reduced ATP synthase (complex V) levels, and consequently impaired ATP production. It has previously been reported that CHCHD3 KD in HeLa cells resulted in fragmented mitochondria that was due to improper mitochondrial fusion. It has also been demonstrated in yeast that individual or combinatorial loss of MICOS complex proteins disrupt cristae morphology, thus suggesting a mechanism by which CHCHD3/6 loss could mediate HLHS pathogenesis. Furthermore, a genetic interaction between was identified SAMM50 and CHCHD3/6 that leads to a contractile deficit and diminished sarcomeric F-Actin. Recent findings demonstrate that SAMM50 directly interacts mammalian CHCHD3, to mediate inner and outer membrane bridging and cristae morphology (Birker, 2023).
The data further suggest that ETC Complex V/ ATP synthase is a potential downstream effector of CHCHD3/6 and MICOS complex function. Individual KD of ATP synthase subunits resulted in reduced fractional shortening and reduced sarcomeric actin. As a result, reduced CHCHD3/6 expression is hypothesized to affects ETC function, specifically ATP synthase, leading to reduced ATP production. Since the MICOS complex is in involved ETC assembly in cristae, ATPase subunits may not be assembled correctly causing mitochondrial dysfunction, accompanied by reduced/abnormal mito-GFP staining. OXPHOS complex assembly has been shown to be disrupted upon MICOS depletion, and it is speculated that ATP synthase function may be disrupted when CHCHD3/6 is reduced. Consistent with this, depletion of ATP synthase levels was observed upon Chchd3/6 KD (Birker, 2023).
Finally, a potential oligogenic basis of HLHS was tested in the
family-based CHCHD3 and CHCHD6 interaction screen and identified three hits that reduced fractional shortening only in conjunction with CHCHD3/6, but not on their own. Co-KD of Cdk12 and Chchd3/6 also reduced fractional shortening, and caused greater lethality relative to Cdk12 KD alone. Cdk12 activates RNA polymerase II to regulate transcription elongation. It was postulated that since Chchd3/6 is a nuclear-encoded gene, reducing transcription with Cdk12 KD could decrease CHCHD3/6 levels in a background where CHCHD3/6 activity is already compromised. Alternatively, reduced transcription of other nuclear genes associated with ATP production in combination with Chchd3/6 KD could further reduce ATP levels enough to cause contractility defects. In support of this, a study examining the effects of RMP (RNA polymerase II subunit 5-mediating protein) found that mice with cardiac-specific Rpm KO exhibited reduced fractional shortening and ATP levels, which were attributed to a reduction in mRNA and protein levels of the mitochondrial biogenesis factor PGC1α. The second hit, goliath, is an endosomal ubiquitin E3 ligase. Although goliath has been implicated in endosomal recycling, its role in Drosophila mitophagy in vivo has not been examined. Reduced cardiac contractility with co-KD of gol and Chchd3/6 could result from impaired mitophagy and reduced mitochondrial biogenesis. Together, the accumulation of damaged mitochondria can reduce ATP content required for contraction. The third hit, β-Spectrin, acts as a scaffolding protein. Recent data suggests that the human ortholog, SPTBN1 (Nonerythroid spectrin β) influences SPTAN1 (Nonerythroid spectrin α) levels, which has a calmodulin binding domain. Therefore, decreased β-Spec expression could reduce Calmodulin levels, thereby reducing contractility due to the combined reduction in Ca2+ handling and Chchd3/6 KD-induced reduced ATP levels (Birker, 2023).
In summary, this study has identified a novel mechanism potentially involved HLHS pathogenesis, starting by analyzing WGS data from a prioritized family and large cohort of HLHS patients, followed by functional testing in vivo using the Drosophila heart model and in vitro using human iPSC-derived CMs. Compromised contractile capacity, diminished sarcomeric F-Actin and Myosin accumulation, and mitochondrial dysfunction in Chchd3/6 KD Drosophila hearts are promising phenotypes that could contribute to early HLHS manifestations or heart failure complications later in life. Further examination of the interactions between the MICOS complex and other emerging candidate genes will identify novel gene functions and pathways that contribute to HLHS pathogenesis. Furthermore, a detailed elucidation of novel candidate genes and genetic interactions based on patient-specific rare potentially damaging variants is expected to lead to gene networks that are relevant for HLHS and other CHDs (Birker, 2023).
Mitochondrial calcium plays critical roles in diverse cellular processes ranging from energy metabolism to cell death. Previous studies have demonstrated that mitochondrial calcium uptake is mainly mediated by the mitochondrial calcium uniporter (MCU) complex. However, the roles of the MCU complex in calcium transport, signaling, and dysregulation by oxidative stress still remain unclear. This study confirmed that Drosophila MCU contains evolutionarily conserved structures and requires essential MCU regulator (EMRE) for its calcium channel activities. Drosophila MCU loss-of-function mutants, which lacked mitochondrial calcium uptake in response to caffeine stimulation, were generated. Basal metabolic activities were not significantly affected in these MCU mutants as observed in examinations of body weight, food intake, body sugar level, and starvation-induced autophagy. However, oxidative stress-induced increases in mitochondrial calcium, mitochondrial membrane potential depolarization, and cell death were prevented in these mutants. It was also found that inositol 1,4,5-trisphosphate receptor (IP3R) genetically interacts with Drosophila MCU and effectively modulates mitochondrial calcium uptake upon oxidative stress. Taken together, these results support the idea that Drosophila MCU is responsible for ER-to-mitochondrial calcium transfer and for cell death due to mitochondrial dysfunction under oxidative stress (Choi, 2017).
Prolonged cellular activity may overload cell function, leading to high rates of protein synthesis and accumulation of misfolded or unassembled proteins, which cause endoplasmic reticulum (ER) stress and activate the unfolded protein response (UPR) to re-establish normal protein homeostasis. Previous molecular work has demonstrated that sleep deprivation (SD) leads to ER stress in neurons, with a number of ER-specific proteins being upregulated to maintain optimal cellular proteostasis. This study investigated the transcriptional and ultrastructural ER and mitochondrial modifications induced by sleep loss. Gene expression analysis in mouse forebrains was used to show that SD was associated with significant transcriptional modifications of genes involved in ER stress but also in ER-mitochondria interaction, calcium homeostasis, and mitochondrial respiratory activity. Using electron microscopy, it was also shown that SD was associated with a general increase in the density of ER cisternae in pyramidal neurons of the motor cortex. Moreover, ER cisternae established new contact sites with mitochondria, the so-called mitochondria associated membranes (MAMs), important hubs for molecule shuttling, such as calcium and lipids, and for the modulation of ATP production and redox state. Finally, it was demonstrated that Drosophila male mutant flies (elav > linker), in which the number of MAMs had been genetically increased, showed a reduction in the amount and consolidation of sleep without alterations in the homeostatic sleep response to SD. This study has provided evidence that sleep loss induces ER stress characterized by increased crosstalk between ER and mitochondria. MAMs formation associated with SD could represent a key phenomenon for the modulation of multiple cellular processes that ensure appropriate responses to increased cell metabolism. In addition, MAMs establishment may play a role in the regulation of sleep under baseline conditions (Aboufares El Alaoui, 2023).
Under stress conditions, mitochondria release low levels of reactive oxygen species (ROS), which triggers a cytoprotective response, called "mitohormesis". It still remains unclear how mitochondria respond to stress-derived stimuli and release a low level of ROS. This study shows that N-acetyl-l-tyrosine (NAT) functions as a plausible intrinsic factor responsible for these tasks in stressed animals. NAT is present in the blood or hemolymph of healthy animals, and its concentrations increase in response to heat stress. Pretreatment with NAT significantly increases the stress tolerance of tested insects and mice. Analyses using Drosophila larvae and cultured cells demonstrate that the hormetic effects are triggered by transient NAT-induced perturbation of mitochondria, which causes a small increase in ROS production and leads to sequential retrograde responses: NAT-dependent FoxO activation increases in the gene expression of antioxidant enzymes and Keap1. Moreover, NAT represses tumor growth, possibly via the activation of Keap1. In sum, it is proposed that NAT is a vital endogenous molecule that could serve as a triggering factor for mitohormesis.
The clearance of mitochondria by autophagy, mitophagy, is important for cell and organism health, and known to be regulated by ubiquitin. During Drosophila intestine development, cells undergo a dramatic reduction in cell size and clearance of mitochondria that depends on autophagy, the E1 ubiquitin-activating enzyme Uba1, and ubiquitin. This study screen a collection of putative ubiquitin-binding domain-encoding genes for cell size reduction and autophagy phenotypes. The endosomal sorting complex required for transport (ESCRT) components TSG101 and Vps36, as well as the novel gene Vps13D were identified. Vps13D is an essential gene that is necessary for autophagy, mitochondrial size, and mitochondrial clearance in Drosophila. Interestingly, a similar mitochondrial phenotype is observed in VPS13D mutant human cells. The ubiquitin-associated (UBA) domain of Vps13D binds K63 ubiquitin chains, and mutants lacking the UBA domain have defects in mitochondrial size and clearance and exhibit semi-lethality, highlighting the importance of Vps13D ubiquitin binding in both mitochondrial health and development. VPS13D mutant cells possess phosphorylated DRP1 and mitochondrial fission factor (MFF) as well as DRP1 association with mitochondria, suggesting that VPS13D functions downstream of these known regulators of mitochondrial fission. In addition, the large Vps13D mitochondrial and cell size phenotypes are suppressed by decreased mitochondrial fusion gene function. Thus, these results provide a previously unknown link between ubiquitin, mitochondrial size regulation, and autophagy (Anding, 2018).
Mitochondrial autophagy or mitophagy is a key process that allows selective sequestration and degradation of dysfunctional mitochondria to prevent excessive reactive oxygen species, and activation of cell death. Recent studies revealed that ubiquitin-proteasome complex activity and mitochondrial membrane rupture are key steps preceding mitophagy, in combination with the ubiquitination of specific outer mitochondrial membrane (OMM) proteins. The deubiquitinating enzyme ubiquitin-specific peptidase 14 (USP14) has been shown to modulate both proteasome activity and autophagy. This study reports that genetic and pharmacological inhibition of USP14 promotes mitophagy, which occurs in the absence of the well-characterised mediators of mitophagy, PINK1 and Parkin. Critical to USP14-induced mitophagy is the exposure of the LC3 receptor Prohibitin 2 by mitochondrial fragmentation and mitochondrial membrane rupture. Genetic or pharmacological inhibition of USP14 in vivo corrected mitochondrial dysfunction and locomotion behaviour of PINK1/Parkin mutant Drosophila model of Parkinson's disease, an age-related progressive neurodegenerative disorder that is correlated with diminished mitochondrial quality control. This study identifies a novel therapeutic target that ameliorates mitochondrial dysfunction and in vivo PD-related symptoms (Chakraborty, 2018).
The mitochondrial contact site and cristae organizing system (MICOS) is a multi-protein interaction hub that helps define mitochondrial ultrastructure. While the functional importance of MICOS is mostly characterized in yeast and mammalian cells in culture, the contributions of MICOS to tissue homeostasis in vivo remain further elucidation. This study examined how knocking down expression of Drosophila MICOS genes affects mitochondrial function and muscle tissue homeostasis. CG5903/MIC26-MIC27 was found to colocalize and function with Mitofilin/MIC60 and QIL1/MIC13 as a Drosophila MICOS component; knocking down expression of any of these three genes predictably altered mitochondrial morphology, causing loss of cristae junctions, and disruption of cristae packing. Furthermore, the knockdown flies exhibited low mitochondrial membrane potential, fusion/fission imbalances, increased mitophagy, and limited cell death. Reductions in climbing ability indicated deficits in muscle function. Knocking down MICOS genes also caused reduced mtDNA content and fragmented mitochondrial nucleoid structure in Drosophila. Together, these data demonstrate an essential role of Drosophila MICOS in maintaining proper homeostasis of mitochondrial structure and function to promote the function of muscle tissue (Wang, 2020).
The eukaryotic porin, also called the Voltage Dependent Anion-selective Channel (VDAC), is the main pore-forming protein of the outer mitochondrial membrane. In Drosophila melanogaster, a cluster of genes evolutionarily linked to VDAC is present on chromosome 2L. The main VDAC isoform, called VDAC1 (Porin1), is expressed from the first gene of the cluster. The porin1 gene produces two splice variants, 1A-VDAC and 1B-VDAC, with the same coding sequence but different 5' untranslated regions (UTRs). The influence of the two 5' UTRs, 1A-5' UTR and 1B-5' UTR, was studied on transcription and translation of VDAC1 mRNAs. In porin-less yeast cells, transformation with a construct carrying 1A-VDAC results in the expression of the corresponding protein and in complementation of a defective cell phenotype, whereas the 1B-VDAC sequence actively represses VDAC expression. Identical results were obtained using constructs containing the two 5' UTRs upstream of the GFP reporter. A short region of 15 nucleotides in the 1B-5' UTR should be able to pair with an exposed helix of 18S ribosomal RNA (rRNA), and this interaction could be involved in the translational repression. These data suggest that contacts between the 5' UTR and 18S rRNA sequences could modulate the translation of Drosophila 1B-VDAC mRNA. The evolutionary significance of this finding is discussed (Leggio, 2018).
The voltage-dependent anion-selective channel (VDAC), also known as mitochondrial porin, is the most abundant protein found in the outer mitochondrial membrane of all eukaryotes. VDAC is the main gateway for the entry and exit of mitochondrial metabolites, and thus it is suspected to control energetic exchanges between the mitochondria and the rest of the cell. Porin/VDAC interacts with several kinases or structural proteins of the cytoskeleton, such as microtubule-associated protein, tubulin and the dynein light chain Tctex-1. Furthermore, porin/VDAC mediates the Ca2+ traffic in the mitochondria. The role of VDAC in cancer is well known. VDAC1 contributes to the phenotype of cancer cells, regulating cellular energy production and metabolism. Indeed, this protein is overexpressed in many cancer types, and silencing of VDAC1 expression induces an inhibition of tumour development. Among others, VDAC1 controls, together with other proteins, the release of the pro-apoptotic factors from mitochondria, e.g. cytochrome c. VDAC1 can also regulate mitochondria-mediated apoptosis by interacting with hexokinases I and II and with proteins of the Bcl2 family, some of which are also highly expressed in many cancers. Moreover, the involvement of VDAC1 in many neurodegenerative diseases, such as amyotrophic lateral sclerosis, Parkinson's, Huntington's25 and Alzheimer's, has also been widely proven (Leggio, 2018).
Lower and higher eukaryotic cells express different sets of porin. In the budding yeast, Saccharomyces cerevisiae, two different porin genes have been identified, POR1 and POR2. In higher eukaryotes, like the mouse and the human, three porin/VDAC proteins are expressed. The structures of mouse and human VDAC1 and that of zebrafish VDAC2 have been resolved and found to exhibit a folding pattern that is similar overall. In contrast, there is a general consensus that each of the VDAC isoforms has distinct physiological roles, because they could specifically interact with different proteins or be differentially sensitive to oxidation by reactive oxygen species (Leggio, 2018).
In Drosophila melanogaster, the genomic locus 2L 32B displays a cluster of four spatially close genes that are evolutionarily linked to VDAC35. These genes share the same exon-intron organization and are very likely to be the result of gene duplication events. However, the main known protein is the product of the first gene (De Pinto, 1989). The second gene in the sequence, porin2, was shown to be expressed in vivo (by histological stainings) and to be able to form permeable pores (using the recombinant protein) (Aiello, 2004). The porin1 gene, which produces VDAC1, is encoded by two main transcripts, 1A-VDAC1 and 1B-VDAC1. These transcripts show two alternative 5' untranslated regions (UTRs) (corresponding to alternative exons 1A or 1B) but the same protein-encoding open reading frame (ORF) and the same 3' UTR sequence ending at one of three different alternative polyadenylation sites. The ORF is for VDAC1, the pore-forming protein of the fly. Thus, in the fly, two alternative splice variants (transcripts) are expressed and are present at all fly development stages and in the same tissues, at the same time, but 1A-VDAC is more abundant (Oliva, 1998). VDAC1 from D. melanogaster has been purified, and its gene has been cloned, sequenced and mapped. This protein shows very conserved functional features (De Pinto, 1998), and it is indeed about 60% identical to the VDAC mammalian isoforms (Leggio, 2018).
The presence of alternative 5' UTRs raised interest and was further investigated. Drosophila transposable P elements, when inserted into the porin locus, abolished VDAC expression and were found to produce a lethal phenotype35. Imprecise excision of such P elements showed that deletion of exon 1B and of its flanking sequences apparently has no effect on normal fly development or on VDAC protein level, whereas deletion of exon 1A suppresses VDAC protein expression. It was therefore suggested that 1B-VDAC could be an unproductive transcript (Leggio, 2018).
This study investigated the function of the two alternative Drosophila 5' UTRs. Overall, the current results suggest that a specific mechanism could be involved in the 1B-VDAC translational regulation. The biological significance of this mechanism is discussed (Leggio, 2018).
This work focused on the regulation of expression of VDAC1 in D. melanogaster. In this species, the porin1 gene produces two alternative transcripts named 1A-VDAC and 1B-VDAC, containing an identical coding sequence but two completely different 5' UTRs. To gain further insights into the biological function of these two alternative splicing forms of VDAC, they were introduced into a VDAC-lacking system, an established S. cerevisiae strain where the porin1 gene was inactivated (δpor1 strain). The advantage of the yeast cell is its viability (under fermentative conditions), whereas D. melanogaster cells cannot survive the deletion of the VDAC1 gene (Leggio, 2018).
In δpor1 yeast, the heterologous 1A-5' UTR directed transcription and translation of VDAC and of GFP used as a reporter; in contrast, the 1B-5' UTR directed the transcription but not the translation of the VDAC or the reporter gene. These results confirm that only the 1A-VDAC, but not the 1B-VDAC, is able to complement the growth defect of the δpor1 yeast cells. Similar data were obtained in Drosophila cells by using a luciferase reporter gene downstream of the 1A- or 1B-5' UTR. These results suggest that the 1B-5' UTR affects VDAC expression by inhibiting protein translation. Furthermore, the results suggest that this mechanism is independent of the coding region cloned downstream of the 5'-UTR (Leggio, 2018).
This study aimed to understand the mechanism responsible for the negative influence of the 1B-5' UTR on the translation of the coding sequences fused downstream. Gene expression in eukaryotic cells is regulated at multiple levels, including mRNA translation. Such control allows rapid changes in protein concentrations and, thus, it is used to maintain cellular homeostasis. Most translation regulation is exerted at the very first stage, when the AUG start codon is identified after the 5' UTR ribosome scanning. Consequently, any occurrence that prevents or inhibits the ability of the ribosome to scan the 5' UTR reduces the efficiency of translation initiation. Mechanisms that produce this effect are well known. Therefore, some of these were assayed, such as the presence of uORFs or stable secondary structures and the association with regulatory RBPs (Leggio, 2018).
The possibility was ruled out that the small upstream ORF (uORF) located in the 1B sequence is involved in translational control. Bioinformatic analysis suggested that no putative strong secondary structure in the untranslated region of 1B-VDAC mRNA should be involved in the inhibition of translation. In addition, bioinformatic predictive analysis of RBPs showed that there is no known RBP specific for the 1B-5' UTR, within the limitations of computational tools. Moreover, the possible involvement of miRNAs was not considered, because the 3' UTR of 1B-VDAC is included in the corresponding 3' UTR of 1A-VDAC, which is longer. Therefore, because a regulatory mechanism involving a miRNA action targeted to this region of 1B-VDAC mRNA could not be specific for the 1B-mRNA, this mechanism was ruled out (Leggio, 2018).
Using a mutagenesis scanning approach, the 16-31 nucleotide region of the 1B-5' UTR sequence was identified as responsible in yeast for the inhibitory effect on translation. The defect in the growth of the δpor1 yeast strain was indeed complemented when the strain was transformed with 1B(Δ16-31)-VDAC mutant, underlining that its removal is sufficient to re-establish the translation. It was also verified that the 16-31 sequence works similarly in Drosophila, although the translation inhibition must rely also on others factors. Therefore, by MS analysis of the proteins bound to an RNA oligo containing the 10-34 sequence of the 1B-5' UTR, proteins were sought that were directly or indirectly involved in the translation control. In particular, eIF4A, eIF5a and Asc1 were recognized. eIF4A is a RNA helicase working in the first stage of translation as a subunit of the cap-binding complex eIF4F, which unwinds the RNA secondary structures in the 5' UTR. Asc1/RACK1 associates with the 40S subunit close to the mRNA exit channel, where it interacts with eIF4E of eIF4F51. Asc1/RACK1 is involved in the control of the translation of housekeeping genes and, in general, represses gene expression. It is known that RACK1 loss-of-function mutations cause early developmental lethality in the mouse and the fly, like VDAC knockout organisms. Moreover, in yeast, loss of ASC1 reduces translation of mitochondrial r-proteins and, like for lack of VDAC1, causes cells to be unable to use non-fermentable carbon sources, demonstrating a direct control of ASC1 on mitochondria functionality. Interestingly, RACK1 has many interaction partners, ranging from kinases and signalling proteins to membrane-bound receptors and ion channels. Thus, under stress conditions, RACK1 can function as a signalling hub of newly synthesised proteins (Leggio, 2018).
From this viewpoint, it can be hypothesised that in yeast the 16-31 sequence might prevents eIF4A function, maybe trapping eIF4A in an inactive conformation. In Drosophila, 1B-VDAC translation could be repressed at the starting point by the coordinated action of more molecules, probably recruited in situ by RACK1. Gus1, which together with Arc1, is known to form a protein complex operating in the control of translation, was identified. In addition, the presence of two different heat-shock proteins (Hsp12 and Hsp76) in this pool of interacting proteins should indicate their recruitment after stress conditions (Leggio, 2018).
The ability of the 1A- and 1B-5' UTR sequences to contact protein-free domains of 18S rRNA, the only rRNA in the 40S subunit, was also tested. Because 18S rRNA mutations impair the integrity of the scanning-competent pre-initiation complex and/or its joining together with the 60S subunit, the translation initiation rate might be reduced by strong and long-range interactions between the protein-free domains of 18S rRNA and the 5' UTR(s) of the incoming mRNA. It has already been demonstrated in eukaryotes that gene expression regulation at the level of translation may occur thanks to specific interactions between mRNAs and rRNA domains. In particular, a highly specific sequence complementarity between 18S rRNA and the 5' UTRs of mRNAs across species has been predicted; this complementarity may modulate the scanning processivity of the 40S subunit through the 5' UTR of mRNAs, which could even stall the initiating PICs in the case of long-range interactions (Leggio, 2018).
In particular, by prediction analysis of RNA:RNA interactions between yeast 18S rRNA and the two alternative D. melanogaster VDAC mRNAs (1A-VDAC and 1B-VDAC), it was found that, in yeast as in D. melanogaster, almost the whole 1B-5'UTR sequence is able to strongly interact with a long sequence of 18S rRNA. In contrast, the 1B(Δ16-31)-5'UTR sequence can only weakly interact with a short sequence of rRNA in the 40S subunit, thus showing a behaviour similar to that 1A-5' UTR. These results underline the relevance of 1B-5' UTR and, in particular in yeast, of its 16-31 sequence for the mechanism of translation control. Interestingly, it was also found that some regions of the rRNA sequence involved in the interaction with the 1B-5' UTR fold in solvent-exposed domains, and some of them are turned towards the mRNA path of the ribosome 40S subunit. Therefore, these rRNA domains should be able to contact the 5' UTR in the incoming 1B-VDAC mRNA, producing a stop in the ribosome scanning. It is noteworthy that a sequence of about 35 nucleotides can be allocated inside the ribosomal mRNA path of PIC and that it was found that almost the whole 1B-5' UTR sequence, (2-116 nucleotides), may potentially interact with three 18S rRNA helices (helix 35, helix 36 and a portion of the helix 34) arranged near the mRNA path at the neck of 40S. In addition, the large helix 33, together with parts of helix 31 and helix 32, being arranged at the beck of the 40S subunit, could easily interact with the 1B-5' UTR. In this way, the 1B-VDAC mRNA translation rate would be negatively controlled by its 5' UTR sequence through the collective action of several interactions with 18S rRNA, the result of which would be a strong delay in ribosome scanning of 1B-VDAC. Probably, this effect in Drosophila could also be the result of additional interactions with fly-specific proteins, ribosomal or not. In any case, it is extremely relevant that the sequences encompassing these rRNA helices are highly conserved between S. cerevisiae and D. melanogaster; this indicates that the mechanism described in the mixed yeast-fly system is likely to act in D. melanogaster (Leggio, 2018).
VDAC is an essential but dangerous protein. Its function as a pro-apoptotic factor is well known and therefore it is essential for the cell to implement a suitable control of VDAC protein level. Also, specific conditions of cell growth involving high energy demand are known to induce up-regulation of VDAC associated with the requirement of mitochondrial biogenesis. Furthermore, these events must be coordinated with the expression of the other mitochondrial proteins, codified by the nuclear genome and from mitochondrial DNA. Therefore, it is conceivable to suppose the presence in the cell of a 'sentry' molecule able to sense, directly or indirectly, the amount of this crucial protein. It was demonstrated that in Drosophila the level of 1B-VDAC transcript is highly increased as a result of overexpression of 1A-VDAC mRNA. When the level of the 1B-VDAC transcript was increased by its overexpression, the endogenous 1A-VDAC mRNA level was meaningfully reduced. Importantly, the results show that the unproductive 1B-VDAC mRNA is able to respond to 1A-VDAC transcript levels, and thus it might work as a molecule signalling the need for activation of mitochondrial biogenesis. This hypothetical role of 1B-VDAC mRNA is supported by its interaction with Asc1/RACK1. Asc1/RACK1 responds to multiple signals, and might act to coordinate the expression of other mitochondrial proteins and thus affect cell respiration (Leggio, 2018).
In addition, the assignment of this important role to 1B-VDAC mRNA might lead to an understanding of why the evolution of the Drosophila genus proceeded towards the acquisition of an alternative 5' UTR with specific features (Leggio, 2018).
In conclusion, these results extend earlier reports and provide further evidence that in D. melanogaster the 1A-VDAC transcript is responsible for protein expression, while the alternative 1B-VDAC mRNA is not active in this respect. Moreover, this work showed that a specific mechanism could be responsible for the translation inhibition of the alternative D. melanogaster 1B-VDAC1 transcript (Leggio, 2018).
The destruction of mitochondria through macroautophagy (autophagy) has been recognised as a major route of mitochondrial protein degradation. Autophagy was originally thought to degrade all mitochondrial proteins at the same rate, but recent work suggests that mitochondrial autophagy may be protein selective. To investigate these questions, a proteomics-based approach was used in the fruit fly Drosophila melanogaster, comparing mitochondrial protein turnover rates in autophagy-deficient Atg7 mutants and controls. ~35% of mitochondrial protein turnover occurred via autophagy. Similar analyses using parkin mutants revealed that parkin-dependent mitophagy accounted for ~25% of mitochondrial protein turnover, suggesting that most mitochondrial autophagy specifically eliminates dysfunctional mitochondria. The results were incompatible with uniform autophagic turnover of mitochondrial proteins and consistent with protein-selective autophagy. In particular, the autophagic turnover rates of individual mitochondrial proteins varied widely, and only a small amount of the variation could be attributed to tissue differences in mitochondrial composition and autophagy rate. Furthermore, analyses comparing autophagy-deficient and control human fibroblasts revealed diverse autophagy-dependent turnover rates even in homogeneous cells. In summary, this work indicates that autophagy acts selectively on mitochondrial proteins, and that most mitochondrial protein turnover occurs through non-autophagic processes (Vincow, 2019).
Decreased cognitive performance is a hallmark of brain aging, but the underlying mechanisms and potential therapeutic avenues remain poorly understood. Recent studies have revealed health-protective and lifespan-extending effects of dietary spermidine, a natural autophagy-promoting polyamine. This study shows that dietary spermidine passes the blood-brain barrier in mice and increases hippocampal eIF5A hypusination and mitochondrial function. Spermidine feeding in aged mice affects behavior in homecage environment tasks, improves spatial learning, and increases hippocampal respiratory competence. In a Drosophila aging model, spermidine boosts mitochondrial respiratory capacity, an effect that requires the autophagy regulator Atg7 and the mitophagy mediators Parkin and Pink1. Neuron-specific Pink1 knockdown abolishes spermidine-induced improvement of olfactory associative learning. This suggests that the maintenance of mitochondrial and autophagic function is essential for enhanced cognition by spermidine feeding. Finally, this study showed large-scale prospective data linking higher dietary spermidine intake with a reduced risk for cognitive impairment in humans (Schroeder, 2021).
Aneuploidy, an unbalanced number of chromosomes, is highly deleterious at the cellular level and leads to senescence, a stress-induced response characterized by permanent cell-cycle arrest and a well-defined associated secretory phenotype. This study used a Drosophila epithelial model to delineate the pathway that leads to the induction of senescence as a consequence of the acquisition of an aneuploid karyotype. Whereas aneuploidy induces, as a result of gene dosage imbalance, proteotoxic stress and activation of the major protein quality control mechanisms, near-saturation functioning of autophagy leads to compromised mitophagy, accumulation of dysfunctional mitochondria, and the production of radical oxygen species (ROS). This study uncovered a role of c-Jun N-terminal kinase (JNK) in driving senescence as a consequence of dysfunctional mitochondria and ROS. Activation of the major protein quality control mechanisms and mitophagy dampens the deleterious effects of aneuploidy, and this study has identified a role of senescence in proteostasis and compensatory proliferation for tissue repair (Joy, 2021).
Mitophagy removes defective mitochondria via lysosomal elimination. Increased mitophagy coincides with metabolic reprogramming, yet it remains unknown whether mitophagy is a cause or consequence of such state changes. The signalling pathways that integrate with mitophagy to sustain cell and tissue integrity also remain poorly defined. Temporal metabolomics was performed on mammalian cells treated with deferiprone, a therapeutic iron chelator that stimulates PINK1/PARKIN-independent mitophagy. Iron depletion profoundly rewired the metabolome, hallmarked by remodelling of lipid metabolism within minutes of treatment. DGAT1-dependent lipid droplet biosynthesis occurred several hours before mitochondrial clearance, with lipid droplets bordering mitochondria upon iron chelation. DGAT1 inhibition restricts mitophagy in vitro, with impaired lysosomal homeostasis and cell viability. Importantly, genetic depletion of DGAT1 in vivo significantly impaired neuronal mitophagy and locomotor function in Drosophila. These data define iron depletion as a potent signal that rapidly reshapes metabolism and establishes an unexpected synergy between lipid homeostasis and mitophagy that safeguards cell and tissue integrity (Long, 2021).
Parkinson's disease-related proteins, PINK1 and Parkin, act in a common pathway to maintain mitochondrial quality control. While the PINK1-Parkin pathway can promote autophagic mitochondrial turnover (mitophagy) following mitochondrial toxification in cell culture, alternative quality control pathways are suggested. To analyse the mechanisms by which the PINK1-Parkin pathway operates in vivo, methods were developed to detect Ser65-phosphorylated ubiquitin (pS65-Ub) in Drosophila. Exposure to the oxidant paraquat led to robust, Pink1-dependent pS65-Ub production, while pS65-Ub accumulates in unstimulated parkin-null flies, consistent with blocked degradation. Additionally, it was shown that pS65-Ub specifically accumulates on disrupted mitochondria in vivo. Depletion of the core autophagy proteins Atg1, Atg5 and Atg8a did not cause pS65-Ub accumulation to the same extent as loss of parkin, and overexpression of parkin promoted turnover of both basal and paraquat-induced pS65-Ub in an Atg5-null background. Thus, this study has established that pS65-Ub immunodetection can be used to analyse Pink1-Parkin function in vivo as an alternative to reporter constructs. Moreover, the findings suggest that the Pink1-Parkin pathway can promote mitochondrial turnover independently of canonical autophagy in vivo (Usher, 2023).
Intronic polymorphic TOMM40 variants increasing TOMM40 mRNA expression are strongly correlated to late onset Alzheimer's Disease. The gene product, hTomm40, encoded in the APOE gene cluster, is a core component of TOM, the translocase that imports nascent proteins across the mitochondrial outer membrane. This study used Drosophila melanogaster eyes as an in vivo model to investigate the relationship between elevated Tom40 (the Drosophila homologue of hTomm40) expression and neurodegeneration. Evidence is provided that an overabundance of Tom40 in mitochondria invokes caspase-dependent cell death in a dose-dependent manner, leading to degeneration of the primarily neuronal eye tissue. Degeneration is contingent on the availability of co-assembling TOM components, indicating that an increase in assembled TOM is the factor that triggers apoptosis and degeneration in a neural setting. Eye death is not contingent on inner membrane translocase components, suggesting it is unlikely to be a direct consequence of impaired import. Another effect of heightened Tom40 expression is upregulation and co-association of a mitochondrial oxidative stress biomarker, DmHsp22, implicated in extension of lifespan, providing new insight into the balance between cell survival and death. Activation of regulated death pathways, culminating in eye degeneration, suggests a possible causal route from TOMM40 polymorphisms to neurodegenerative disease (Periasamy, 2022).
Following acute genotoxic stress, both normal and tumorous stem cells can undergo cell-cycle arrest to avoid apoptosis and later re-enter the cell cycle to regenerate daughter cells. However, the mechanism of protective, reversible proliferative arrest, "quiescence," remains unresolved. This study shows that mitophagy is a prerequisite for reversible quiescence in both irradiated Drosophila germline stem cells (GSCs) and human induced pluripotent stem cells (hiPSCs). In GSCs, mitofission (Drp1) or mitophagy (Pink1/Parkin) genes are essential to enter quiescence, whereas mitochondrial biogenesis (PGC1α) or fusion (Mfn2) genes are crucial for exiting quiescence. Furthermore, mitophagy-dependent quiescence lies downstream of mTOR- and PRC2-mediated repression and relies on the mitochondrial pool of cyclin E. Mitophagy-dependent reduction of cyclin E in GSCs and in hiPSCs during mTOR inhibition prevents the usual G1/S transition, pushing the cells toward reversible quiescence (G0). This alternative method of G1/S control may present new opportunities for therapeutic purposes (Taslim, 2023).
Mitochondrial malfunction and autophagy defects are often concurrent phenomena associated with neurodegeneration. This study shows that Miga, a mitochondrial outer-membrane protein that regulates endoplasmic reticulum-mitochondrial contact sites (ERMCSs), is required for autophagy. Loss of Miga results in an accumulation of autophagy markers and substrates, whereas PI3P and Syx17 levels are reduced. Further experiments indicated that the fusion between autophagosomes and lysosomes is defective in Miga mutants. Miga binds to Atg14 and Uvrag; concordantly, Miga overexpression results in Atg14 and Uvrag recruitment to mitochondria. The heightened PI3K activity induced by Miga requires Uvrag, whereas Miga-mediated stabilization of Syx17 is dependent on Atg14. Miga-regulated ERMCSs are critical for PI3P formation but are not essential for the stabilization of Syx17. In summary, this study identified a mitochondrial protein that regulates autophagy by recruiting two alternative components of the PI3K complex present at the ERMCSs (Xu, 2022).
Eukaryotic cells are compartmentalized into different organelles that execute distinct functions and communicate with each other through indirect signal transduction or direct organelle-organelle contacts. Mitochondria and the adjacent endoplasmic reticulum (ER) form contacts, which are characterized by a 10-30 nm distance between the two organelles. These contacts mediate lipid exchange and calcium flux between the ER and mitochondria. It has been reported that ER-mitochondrial contact sites (ERMCSs) are important platforms for regulating macroautophagy (hereafter referred to as autophagy) and mitophagy (Xu, 2022).
Autophagosome formation at the ERMCSs in mammalian cells has been reported. Upon starvation, the ER-resident SNARE protein syntaxin 17 (STX17 in mammals; Syx17 in flies) recruits the PI3K complex subunit Atg14 to the ERMCSs and triggers autophagosome formation. However, Syx17 was not required for autophagosome formation in flies , and the major role of Syx17 in both mammals and flies is to mediate the fusion between autophagosome and lysosome. In addition, VAPB and PTPIP51, a pair of ERMCS tethers, also regulate autophagy. Increased ERMCS formation facilitated by VAPB or PTPIP51 overexpression inhibits autophagy; conversely, the weakening of contact by knockdown of these tethers stimulates autophagosome formation. Recent studies have shown that autophagy occurs at ERMCSs to supply free fatty acids for mitochondrial energy metabolism, while mitochondrial respiratory chain activity supports autophagy through the regulation of ERMCS formation. In addition to regulating autophagy at the initiation stage, in a previous study, it was determined that mitochondria play a crucial role in the late stage of autophagy. The loss of Tom40, a key subunit of the mitochondrial protein import channel, results in blockage of autophagosome and lysosome fusion. It was also found that defects in several general mitochondrial metabolic processes, such as ATP production, mitochondrial protein synthesis, or the citrate cycle, do not cause the autophagy defects observed in Tom40-depleted tissues. This implied that the autophagy defects caused by blocking mitochondrial protein import are rather specific. It is therefore hypothesized that certain mitochondrial proteins regulate autophagy directly (Xu, 2022).
In the present study, it was demonstrated that Miga, a mitochondrial outer-membrane protein, is required for autophagy. Loss of Miga led to defects in autophagosome-lysosome fusion. Miga is an evolutionarily conserved protein, with orthologs from worms to humans. In a previous study, it was found to be localized on the mitochondrial outer membrane to regulate mitochondrial fusion by stabilizing MitoPLD. Miga interacts with the ER-localized VAP protein to establish ERMCSs. The interactions between Miga and VAP proteins are regulated by the phosphorylation of the FFAT motif in Miga. A recent study also reported that MIGA2 (the human ortholog of Miga) regulates ERMCSs and contacts between mitochondria and lipid droplets (LDs) . Loss of Miga led to the degeneration of photoreceptor cells in flies. Overexpression of Miga in fly eyes resulted in increased ERMCSs and severe eye degeneration. In mice, loss of MIGA2 led to anxiety-like behavior. This study found that Miga interacts with Atg14 and Uvrag to regulate PI3K activity and Syx17 stability, thereby modulating autophagy (Xu, 2022).
Defects in both mitochondria and autophagy are hallmarks of several types of neurodegenerative diseases. This study found that Miga establishes a direct link between mitochondria and autophagy to maintain cellular homeostasis (Xu, 2022).
It is striking that a mitochondrial protein directly regulates autophagy by interacting with the core components of the autophagy machinery. In the present study, it was found that the mitochondrial protein Miga forms complexes with Uvrag and Atg14 to regulate PI3P production and to stabilize Syx17 during autophagy (Xu, 2022).
Miga interacts with Vap33 to mediate formation of ERMCSs. Overexpression of wild-type Miga, but not MigaFM, led to increased PI3P levels, implying that Miga-induced ERMCSs are required for regulating PI3P formation. However, the ERMCS tether function of Miga is neither required for recruiting Uvrag nor for binding to Atg14 and Syx17 stabilization. It has been shown previously that Atg14 and other components of the PI3K complex, such as Atg16 and Vps34, are enriched in ERMCSs upon starvation. The question remains as to why the PI3K complex needs to be present. Phosphatidylinositol (PI) is a substrate required for the PI3K complex to produce PI3P. PI is synthesized on the ER, and ERMCSs are the sites for the transfer of PI between the ER and mitochondria. During autophagy, the PI3K complex promotes PI3P formation to facilitate autophagic processes, and ERMCSs represent platforms to access PI. It is believed that the enrichment of the PI3K complex at ERMCSs is needed to assess the supply of PI. The present study found that MigaFM failed to promote PI3P formation, although it was still able to recruit key PI3K components, such as Uvrag or Atg14. This implied that PI3P formation during autophagy not only requires the activity of the PI3K complex but also PI supplied from ERMCSs (Xu, 2022).
Previous studies reported that ERMCSs are required for the initiation of autophagy. In the current study, it was found that in Miga mutants, autophagic processes were blocked at the autophagosome-lysosome fusion stage, while autophagosome formation was largely unaffected. Lack of Miga led to a reduction in PI3P and Syx17 levels. Previous studies have demonstrated that PI3K is not only essential for autophagy initiation but is also recruited to the autophagosome together with the HOPS complex to facilitate autophagosome and lysosome fusion in mammalian cells. The remaining PI3P in Miga mutants is probably sufficient for autophagosome formation but not enough for the autophagosome-lysosome fusion process. This study found that the loss of Miga reduced co-localization of FYVE-GFP and Atg8a but not co-localization of FYVE-GFP and CathL. This suggests that the reduction of PI3P in autophagosomes, but not lysosomes, might contribute to fusion defects (Xu, 2022).
The fusion defects observed in Miga mutants were not identical to those found in mutants without Syx17 or HOPS components. The puncta of autophagosome markers are larger in Miga mutants than those in mutants without Syx17 or HOPS components, possibly due to the combined effects of reduction of PI3P and Syx17. In worms and mammalian cells, the lack of EPG5 prevents autophagosome maturation and induces the ectopic fusion of autophagosomes with various endocytic vesicles. The enlarged Atg8a-positive structures in Miga mutants might also be a result of the ectopic fusion of autophagosomes with other vesicles (Xu, 2022).
In mammals, both UVRAG and ATG14 are required for autophagy. In flies, Uvrag regulates PI3P formation under fed conditions, and Atg14 is required for PI3P-positive autophagosome formation. This study found that Miga overexpression induces PI3P formation; additionally, Uvrag, but not Atg14, is required during this process. It was also found that Miga overexpression leads to an upregulation of numerous autophagy markers, such as Atg9, Syx17, Atg18a, Rab7, and LAMP, among others. However, the expression levels or patterns of p62 and Atg8a did not change significantly upon Miga overexpression. This implied that Miga overexpression is not sufficient to fully activate autophagy (Xu, 2022).
STX17, the mammalian ortholog of Syx17, is an autophagosome-localized Q-SNARE that mediates autophagosome and lysosome fusion through interactions with SNAP29 and VAMP8/Vamp7. STX17 contains two tandem transmembrane domains that have low hydrophobicity but are required for autophagosome localization. In fed mammalian cells, STX17 reportedly localizes to the ER, mitochondria, and cytosol. STX17 was enriched in ERMCSs upon autophagy stimulation and was present on completely closed autophagosomes. The detailed translocation mechanism remains unclear. In flies, Syx17 shows diffusely dispersed patterns, and there is no mitochondria-specific localization under normal fed conditions. Syx17 forms puncta and co-localizes with Atg8-positive autophagosomes upon starvation. It was found that Miga is required for the stabilization of Syx17. Miga does not bind to Syx17 but stabilizes it through Atg14. It is puzzling why a mitochondrial protein would be required for the stabilization of a protein that functions in autophagosome maturation. It has been reported that there are three-way contacts among the ER, mitochondria, and late endosomes. It is possible that Miga, Vap33, and Atg14 mediate the contact between the ER, mitochondria, and autophagosomes. Autophagosome-associated Atg14 further stabilizes Syx17 to mediate the fusion between autophagosomes and lysosomes. GFP-Atg14 formed large puncta instead of decreasing in the Miga mutant clones. One possible explanation for this is that the overexpression of GFP-Atg14 overrides the requirement of Miga to stabilize it, but the overexpression of GFP-Atg14 per se is not sufficient to fully rescue the autophagy defects in the Miga mutant. Therefore, similar to other autophagy markers, GFP-Atg14 puncta accumulated in the Miga mutant clones (Xu, 2022).
In summary, this study identified a mitochondrial protein, Miga, that regulates autophagic processes by interacting with Atg14 and Uvrag. This delineates a link between mitochondria and macroautophagy. However, this study did not solve how Miga stabilizes Atg14 and Syx17. It is possible that Miga mediates the three-way contact between the ER, mitochondria, and autophagosomes. Miga interacts with Atg14 and stabilizes Atg14. Furthermore, Atg14 interacts with Syx17 to stabilize it. It is not clear how the relay is carried out during autophagy (Xu, 2022).
Both Atg14 and Uvrag interact with MigaN (1-252 aa), but there is no evident competition between Atg14 and Uvrag. The exact regions of Miga that bind to each protein were not identified in this study (Xu, 2022).
Cell proliferation and tissue growth depend on the coordinated regulation of multiple signaling molecules and pathways during animal development. Previous studies have linked mitochondrial function and the Hippo signaling pathway in growth control. However, the underlying molecular
mechanisms are not fully understood. This study identifies a Drosophila
mitochondrial inner membrane
protein ChChd3
as a novel regulator for tissue growth during larval development. Loss of ChChd3 leads to
tissue undergrowth and cell proliferation defects. ChChd3 is
required for mitochondrial fusion and removal of ChChd3 increases
mitochondrial fragmentation. ChChd3 is another mitochondrial
target of the Hippo pathway,
although it is only partially required for Hippo pathway mediated overgrowth. Interestingly, lacking of ChChd3 leads to inactivation of Hippo activity under normal development, which is also dependent on the transcriptional co-activator Yorkie (Yki). Furthermore, loss of ChChd3 induces oxidative stress and activates the JNK pathway. In addition, depletion of other mitochondrial fusion components, Opa1 or Marf, inactivates the Hippo pathway as well. Taken together, the study proposes that there is a crosstalk between mitochondrial fusion and the Hippo pathway which is essential in controlling cell proliferation and tissue homeostasis in Drosophila (Deng, 2016).
Mitochondrial fusion and fission affect the distribution and quality control of mitochondria. This study shows that Marf (Mitochondrial associated regulatory factor), is required for mitochondrial fusion and transport in long axons. Moreover, loss of Marf leads to a severe depletion of mitochondria in neuromuscular junctions (NMJs). Marf mutants also fail to maintain proper synaptic transmission at NMJs upon repetitive stimulation, similar to Drp1 fission mutants. However, unlike Drp1, loss of Marf leads to NMJ morphology defects and extended larval lifespan. Marf is required to form contacts between the endoplasmic reticulum and/or lipid droplets (LDs) and for proper storage of cholesterol and ecdysone synthesis in ring glands. Interestingly, human Mitofusin-2 rescues the loss of LD but both Mitofusin-1 and Mitofusin-2 are required for steroid-hormone synthesis. These data show that Marf and Mitofusins share an evolutionarily conserved role in mitochondrial transport, cholesterol ester storage and steroid-hormone synthesis (Sandoval, 2014).
Mitochondrial dynamics plays a critical role in the control of organelle shape, size, number, function and quality control of mitochondria from yeast to mammals. It consists of fusion and fission of mitochondria, which are regulated by several GTPases. Mitochondrial fusion requires the fusion of the outer membrane followed by inner membrane fusion. In mammals, Mitofusin 1 (Mfn1) and Mitofusin 2 (Mfn2) regulate outer mitochondrial fusion whereas inner membrane fusion is controlled by Optic atrophy protein 1 (Opa1). Mitochondrial fission is regulated by Dynamin related protein 1 (Drp1). Decreased fusion results in fragmented round mitochondria, while defective fission leads to fused and enlarged mitochondria (Sandoval, 2014).
Loss of these mitochondrial GTPases results in lethality in worms, flies and mice. Mutations in the human DRP1 gene causes a dominant fatal infantile encephalopathy associated with defective mitochondrial and peroxisomal fission. On the other hand, missense mutations in OPA1 lead to a dominant optic atrophy. Depending on the severity of the mutation, patients may also suffer from ataxia and neuropathy. Also, missense mutations in MFN2 cause Charcot-Marie-Tooth type 2A, a common autosomal dominant peripheral neuropathy associated with axon degeneration. Finally, aberrant levels of mitochondrial GTPases have been associated with Parkinson's, Huntington's and Alzheimers' diseases. These observations in model organisms and human patients suggest that mitochondrial dynamics affects neuronal maintenance in many different contexts (Sandoval and references therein, 2014).
A significant imbalance of mitochondrial fission and fusion may affect the subcellular distribution of mitochondria, especially in neurons since they need to efficiently traffic from the soma to the synapses. Loss of Drosophila Drp1 impairs the delivery of mitochondria to neuromuscular junctions (NMJs), likely because they are large and interconnected. This defect is also associated with a severe depletion of mitochondria in NMJs, which affects local ATP production. This in turn affects the trafficking of synaptic vesicles upon endocytosis during prolonged stimulation. Similarly, in vertebrates, loss of Drp1 leads to an accumulation of mitochondria in the soma and reduced mitochondrial density in dendrites of hippocampal neurons. The Drp1 data in flies and vertebrates indicate that the expanded size of mitochondria affects their mobility (Sandoval, 2014).
Mitochondrial trafficking may also be affected by the physical interaction between the mitochondria and the transport machinery. Recent studies have documented a direct interaction between Mfn2 and a motor adaptor complex for mitochondrial transport, Miro2 (see Drosophila Miro). Moreover, loss of MFN2 in Purkinje cells displayed reduced mitochondrial motility in cerebellar dendrites and reduced mitochondrial transport in axons in cultured dorsal root ganglion neurons. These data suggest that an interaction of Mfn2 with Miro2 may be important for its role in trafficking. Although loss of both Drp1 and MFN2 impair mitochondrial trafficking, a careful comparison of the phenotypes associated with loss of Drosophila Drp1, Mitofusin or Marf, would be useful as the suggested mechanisms by which they impair transport seem very different (Sandoval, 2014).
In addition to their roles in fission and fusion, Drp1, Mfns and Opa1 have been implicated in a variety of other processes. For example, Drp1 has been shown to facilitate the induction of apoptosis whereas Opa1 was shown to affect the stability of cristae junction in inner mitochondrial membrane. Finally, Mfn2 also tethers mitochondria to the endoplasmic reticulum (ER) to mediate Ca2+ uptake (de Brito, 2008). However, the molecular mechanisms underlying these non-canonical functions are less well studied (Sandoval, 2014).
In an unbiased screen designed to identify essential genes that affect neuronal function, the first mutant allelic series was identified of Marf in Drosophila. This study exploits these mutants to determine how loss of Marf affects mitochondrial transport when compared to Drp1 loss. Surprisingly, NMJ defects were identified only in Marf mutants but not in Drp1 mutants. These defects are regulated non-cell autonomously by steroid-hormones produced in ring glands (RG), a major endocrine organ in insects. Through expression of human MFN1 or MFN2 in Marf mutant RG, it was shown that MFN1 and MFN2 have both distinct and complementary roles (Sandoval, 2014).
How does loss of fission or fusion affect mitochondrial function? In the absence of fusion mixing of mitochondrial DNA and proteins may be severely impaired. Given that mitochondrial proteins are in an environment rich in oxygen radicals, lack of fusion may cause more damage than when fission is impaired. Simply stated, loss of fusion proteins like Marf, MFN1 or MFN2 may cause more severe phenotypes than the loss of a fission protein like Drp1. Moreover, proteins like Marf and Drp1 may perform other functions that are not directly related to fusion or fission, and hence affect other processes. Based on a careful phenotypic comparison of loss of Marf and Drp1 in Drosophila many similarities and differences were found (Sandoval, 2014).
Marf mutants display small mitochondria whereas Drp1 mutants exhibit large fused mitochondria. Interestingly, both mutants accumulate mitochondria in the cell body of the neurons and the proximal axonal segments. In Drp1 mutants, the mitochondria seem to be severely elongated in axons where they fail to reach the NMJs, as previously described. The impairment in axonal transport is thought to be due to the fact that the mitochondria are hyperfused and cannot easily be transported. Indeed, loss of Marf in Drp1 mutants can restore mitochondrial trafficking proximally but distal axonal trafficking is still impaired. In Marf mutants, even though mitochondria are small and can enter the axons, the numbers of mitochondria that travel distally toward the NMJs are dramatically reduced. Hence, loss of Marf impairs mitochondrial trafficking and longer axons are more severely affected than shorter axons. Since longer axons are more severely affected in CMT2A patients, defects in mitochondrial trafficking may be at the root of some of the phenotypes associated with the disease (Sandoval, 2014).
Mfn2 has been implicated in axonal transport via binding to Miro2. Indeed, knockdown of MIRO2 in cultured vertebrate neurons affects mitochondrial transport in an identical fashion as loss of MFN2. However, the severity of mitochondrial transport that observed in Marf mutants is much less pronounced than what has been described in dmiro mutants and what was observe when dmiro is lost. Moreover, removal of dmiro in Marf mutants dramatically enhances the Marf phenotype and almost abolishes axonal localization of mitochondria, arguing that Marf cannot be solely responsible for mitochondrial transport in Drosophila (Sandoval, 2014).
A comparison of the presence of mitochondria at NMJ synapses shows that Marf mutants have fewer mitochondria than Drp1 mutants. Moreover, Marf mutants but not Drp1 mutants display a severe increase in small clustered boutons. The small and clustered boutons have also been observed in other mutants like endophilin, synaptojanin, eps15, dap 160, flower and dmiro. However, unlike in Marf mutants, the bouton phenotypes are fully rescued by neuronal expression of the cognate protein within MN in the above mentioned mutants. Moreover, knockdown of Marf in neuron, muscle or glia does not recapitulate the bouton phenotype observe in Marf mutants, suggesting a unique cell non-autonomous requirement of Marf for proper NMJ morphology (Sandoval, 2014).
Marf mutants exhibit two obvious phenotypes at NMJs: a severe depletion of mitochondria and a doubling of the number of boutons combined with a severe reduction in size whereas Drp1 mutants only exhibit a severe reduction in mitochondria. However, electrophysiological studies show that loss of Marf does not affect basal synaptic transmission similar to what is observed in Drp1 mutants. Both respond similarly to wild type NMJs when stimulated at 0.2 Hz and both show a progressive run down at 10 Hz when compared to controls. Moreover, endocytosis using FM1-43 and 60 mM K+ is not impaired in Marf and Drp1 mutants, suggesting a defect in reserve pool mobilization in both mutants. The data also show that the bouton defects observed in Marf mutants do not contribute to the run down in synaptic transmission since Drp1 boutons are normal in number and size yet also have a run down in synaptic transmission (Sandoval, 2014).
Loss of Marf in ring glands (RG) recapitulates the bouton phenotype observed in Marf mutants and expression of Marf in RG fully rescues this phenotype. Interestingly, both Marf and Opa1 are required for steroid hormone production and both lead to extended larval lifespan when knocked down in the RG only (8-10 days), whereas Drp1 mutations do not affect steroid hormone synthesis. Reduction of ecdysone production by knockdown of the prothoracicotropic hormone receptor (torso) in the RG also leads to an extended larval lifespan (9 days) and an increased growth of NMJs. Interestingly, knockdown of Drosophila SUMO (dsmt3) in RG lead to a defect in cholesterol import in the RG, reduced 20E levels and an extended larval lifespan (19 days). Hence, the severe reduction in ecdysone synthesis in Marf mutant RG underlies the prolonged larva stages and NMJ morphological defects (Sandoval, 2014).
The reduction in the number of LDs in RGs when Marf is lost suggests that these RGs are unable to store cholesterol. This storage of cholesterol esters probably permits the RG to produce large amounts of ecdysone when needed, especially at the larval stage and larval to pupal transitions. Cholesterol storage and steroid hormone biosynthesis requires both the ER and mitochondria in vertebrates but loss of MFN1 or MFN2 have not been shown to affect LD synthesis. Defects of anchoring mitochondria to the ER and LDs in Marf RGs argue that these defects lead to the loss of LD and production of ecdysone. In agreement with this hypothesis, expression of human MFN2, which tethers ER to mitochondria, in Marf mutants restores LD synthesis and organelle contacts. Moreover, expression of human MFN2 in RNAi mediated Marf knockdown in neurons and muscles rescues ER morphology and stress. However, MFN2 expression alone in Marf mutant RG did not restore ecydsone synthesis, arguing that there are other mitochondrial defects associated with the loss of Marf (Sandoval, 2014).
The current data show that co-expression of human MFN1 and MFN2 fully rescue the observed phenotypes in Marf mutants. Although RG-specific expression of MFN1 in Marf mutants did not restore LD numbers or organelle contacts, MFN1 is still necessary for ecdysone synthesis together with MFN2, suggesting a role downstream of cholesterol ester storage for both proteins. Moreover, knockdown of Opa1 in RG did not alter LD numbers but causes reduced 20E levels and aberrant NMJs. Opa1 resides within the inner mitochondrial membrane, suggesting its role in ecdysone synthesis is within the mitochondria. Ecdysone synthesis within the mitochondria requires two cytochrome p450 enzymes encoded by disembodied and shadow. Hence, it is likely that impairment in fusion but not fission affects the function of these enzymes (see Model of Marf dual function in steroid synthesis in the ring glands) (Sandoval, 2014).
Opa1 and MFN2 but not Drp1 have been implicated in vertebrate steroidogenesis. Interestingly, in placental trophoblast cells (BeWO) in culture the loss of OPA-1 promotes progesterone production by 70% whereas loss of MFN2 has been reported to lead to a 20% decrease in progesterone production. In contrast, testosterone production in MA-10 Leydig cells was unaffected by loss of OPA1 whereas loss of MFN2 did affect testosterone production by 40% in MA-10 Leydig cells. Hence, in both vertebrate endocrine cells, loss of MFN2 or OPA-1 affected steroids very differently as was observe very similar phenotypes associated with the loss of either protein in the current study. This study also suggests that MFN2 functions upstream of cholesterol entry into the mitochondria at the cholesterol storage stage, since MFN2 restores LD synthesis in Drosophila RG. However, rescuing LD production is not sufficient to restore ecdysone synthesis, suggesting a secondary defect (see Model of Marf dual function in steroid synthesis in the ring glands). In summary, these data indicate that MFN1 and MFN2 have separate functions in vivo that are integrated in a single protein in fly Marf (Sandoval, 2014).
Mitochondrial dynamism (fusion and fission) is responsible for
remodeling interconnected mitochondrial networks in some cell
types. Adult cardiac myocytes lack mitochondrial networks, and
their mitochondria are inherently “fragmented”.
Mitochondrial fusion/fission is so infrequent in cardiomyocytes as
to not be observable under normal conditions, suggesting that
mitochondrial dynamism may be dispensable in this cell type.
However, it has been previously reported that
cardiomyocyte-specific genetic suppression of mitochondrial fusion
factors Optic atrophy 1 (Opa1) and Mitofusin
(Marf) evokes
cardiomyopathy in Drosophila hearts. Fusion-mediated
remodeling of mitochondria may be critical for cardiac
homeostasis, although never directly observed. Alternately, inner
membrane Opa1 and outer membrane mitofusin/MARF might have other
as-yet poorly described roles that affect mitochondrial and
cardiac function. This study compared heart tube function in three
models of mitochondrial fragmentation in Drosophila
cardiomyocytes: Drp1 expression, Opa1 RNAi, and mitofusin MARF
RNA1. Mitochondrial fragmentation evoked by enhanced Drp1-mediated
fission did not adversely impact heart tube function. In contrast,
RNAi-mediated suppression of either Opa1 or mitofusin/MARF induced
cardiac dysfunction associated with mitochondrial depolarization
and ROS production. Inhibiting ROS by overexpressing superoxide
dismutase (SOD) or suppressing ROMO1
prevented mitochondrial and heart tube dysfunction provoked by
Opa1 RNAi, but not by mitofusin/MARF RNAi. In contrast, enhancing
the ability of endoplasmic/sarcoplasmic reticulum to handle stress
by expressing Xbp1
rescued the cardiomyopathy of mitofusin/MARF insufficiency without
improving that caused by Opa1 deficiency. The study concludes that
decreased mitochondrial size is not inherently detrimental to
cardiomyocytes. Rather, preservation of mitochondrial function by
Opa1 located on the inner mitochondrial membrane, and prevention
of ER stress by mitofusin/MARF located on the outer mitochondrial
membrane, are central functions of these “mitochondrial
fusion proteins” (Bhandari, 2015). By performing a side-by-side detailed comparison of
cardiomyopathies provoked by interrupting fusion of either the
outer or inner mitochondrial membranes, this study identified
distinct cellular mechanisms for the different molecular lesions.
RNAi-mediated suppression of outer mitochondrial membrane
mitofusin/MARF and inner mitochondrial membrane Opa1 provokes
similar overt phenotypes: in both models mitochondrial size is
approximately halved, the proportion of depolarized (sick)
mitochondria increases to ~ 40%, mitochondrial ROS production is
comparably greater (~ 50%), and the heart tubes exhibit similar
reductions in fractional shortening. However, the cardiac defect
caused by Opa1 deficiency is readily corrected by attacking the
disease at the level of mitochondrial ROS production, through SOD
expression or ROMO1 suppression. Indeed, mitochondrial structural
and functional abnormalities are also improved by these genetic
maneuvers, suggesting that they interrupt a vicious cycle of
ROS-induced mitochondrial degeneration provoked by Opa1
insufficiency. Thus, interrupting endogenous mitochondrial ROS
production greatly abrogates both the mitotoxicity and the
cytotoxicity evoked by Opa1 suppression. This suggests a central
role for mitochondrial degeneration in the Opa1-deficient fly
heart model and, by extension, other heart diseases caused by
defective inner mitochondrial membrane fusion proteins (Bhandari,
2015). Whereas mitochondrial size and polarization status are similarly
impaired in mitofusin/MARF insufficient heart tubes, neither of
the mitochondrial-targeted interventions directed at reducing ROS,
both of which rescue Opa1 deficient hearts, improve the
cardiomyopathy induced by mitofusin/MARF deficiency. Indeed,
whereas transgenic expression of SOD1 or SOD2 normalizes both ROS
and heart tube function in Parkin-deficient heart tubes and
Opa1-deficient heart tubes, it is remarkable that SOD fails to
improve ROS levels in the mitofusin/MARF deficient heart tubes.
Together with the original observation of transient heart tube
functional improvement with SOD1, these observations suggest that
there is “ROS escape” in the mitochondrial fusion
impaired model that confers resistance to SOD expression or ROMO1
suppression. Instead, heart tube function is normalized without
improving either mitochondrial structural or functional
abnormalities by genetically enhancing the cardiomyocytes' ability
to handle ER stress through XBP1 expression. These results,
although surprising, are consistent with an essential role for
mitofusin-mediated mitochondrial-ER/SR cross-talk in managing the
ER stress response as proposed earlier in heart and in other
tissues (Bhandari, 2015). Previously, the consequences of Opa1 deficiency on Drosophila
eye phenotypes have been found to be rescuable with SOD1, which is
in accordance with findings of this study. Another study in Drosophila
neurons and skeletal muscle also supports an important role for
mitofusins, but not Opa1, as modulators of ER stress. However,
only human Mfn2,
and not human Mfn1,
can correct abnormalities induced by mitofusin/MARF suppression in
flies. This contrasts with findings in this study that
cardiac-specific expression of either human Mfn1 or Mfn2 will
fully correct cardiomyopathy induced by cardiomyocyte-specific
MARF RNAi. Furthermore, ER dysmorphology has been found in
MARF-deficient fly tissues, which was not detected in
MARF-deficient heart tubes. It is likely either that the heart has
different requirements for mitofusins and ER/SR morphology, or
that the powerful tinman
gene promoter used for cardiomyocyte-specific gene manipulation
confers different expression characteristics to the heart models.
Either way, the overall conclusions regarding a role of ER stress
in mitofusin deficiency are in agreement (Bhandari, 2015). Cardiomyocyte mitochondria are the Oompa-Loompas of the heart
(with apologies to Roald Dahl): They are diminutive, structurally
homogenous, and frequently overlooked despite toiling endlessly
behind the scenes to keep the place running. Research has tended
to focus on the mitochondrial work product (cardiac metabolism)
and the means by which the general mitochondrial population is
sustained (through biogenesis), rather than the fate of individual
organelles. Indeed, individual cardiomyocyte mitochondria seem
hardly worthy of observation, being monotonously similar in
appearance and lacking the morphometric remodeling or
intra-cellular mobility that has sparked detailed investigations
(and visually engaging movie clips) in other cell types. The data
presented in this study emphasize that (for mitochondria as well
as Oompa-Loompas) size is not the critical determinant of
function; it is literally what is inside that counts. Accordingly,
one should eschew generalizations and extrapolations of
mitochondrial status and dysfunction based strictly on
morphometry (Bhandari, 2015). Mitochondria are involved in a variety of cellular functions, including ATP production, amino acid and lipid biogenesis and breakdown, signalling and apoptosis. Mitochondrial dysfunction has been linked to neurodegenerative diseases, cancer and ageing. Although transcriptional mechanisms that regulate mitochondrial abundance are known, comparatively little is known about how mitochondrial function is regulated. This study identifies the metabolite stearic acid (C18:0), which was deficient in Elovl6 (Baldspot) mutant flies, To study the function of very long chain fatty acids, this study analyzed Drosophila lacking Elovl6, the enzyme elongating C16 fatty acids to C18. Sequence analysis identified noa8 as fly Elovl6 (dElovl6). On standard laboratory food, dElovl6 loss-of-function animals die as early larvae. This study confirmed dElovl6- mutants have impaired C16:0-C18:0 elongase activity and reduced C18:0 levels, and their lethality is rescued by human Elovl6. Survival to pupation was rescued by supplementing fly food (containing little lipid), with C18:0, but not C18:1 or C20:0, confirming the larval lethality is due to C18:0 deficit (Senyilmaz, 2015).
Removing antifungal agents from fly food improved Elovl6- survival. Since these agents are mitotoxins, this suggested Elovl6- mutants might be hypersensitive to mitochondrial inhibition. Indeed, sub-lethal concentrations of Rotenone, a Complex I inhibitor, killed Elovl6- mutants when added to antifungal-free food, but other drugs did not. Thus mitochondrial function is limiting in Elovl6- mutants. Elovl6- mutants have impaired mitochondrial respiration, rescued by dietary C18:0 supplementation or by expressing an alternative oxidase AOX9 allowing bypass of Complexes III+IV. Complex IV activity was not impaired in Elovl6- mutants, suggesting Elovl6- mutants suffer from a Complex III defect (Senyilmaz, 2015).
If the main cause of Elovl6- lethality is reduced mitochondrial function, then viability should be rescued by restoring mitochondrial functional capacity. Indeed, Elovl6- viability was rescued by expressing AOX or PGC1a, driving mitochondrial biogenesis. Thus the organismal function of C18:0 is less pleiotropic than expected. Interestingly, Drosophila Elovl6 localizes to mitochondrial outer membranes (Senyilmaz, 2015).
Lipidomic analysis of purified mitochondria revealed their membranes have little C18:0, suggesting C18:0 does not play a structural role in mitochondria. Elovl6- mutants also did not have fewer mitochondria than controls (assayed by porin levels and citrate synthase activity. This study therefore investigated if C18:0 regulates mitochondrial activity. Mitochondria dynamically fuse and fission to form tubular structures10,11. Elovl6- mutants had hyper-fragmented mitochondria, rescued by dietary C18:0 supplementation. dElovl6 knockdown in S2 cells reproduced this phenotype, indicating it is cell autonomous. Importantly, S2 cells were grown in Serum Free Medium, which lacks fatty acids normally bound to serum BSA. Mitochondria of dElovl6 knockdown cells rapidly re-fused upon addition of BSA-conjugated C18:0 to the medium for just 120 or 20 minutes. Mitochondria of HeLa cells also fragmented when grown in medium with serum that was delipidated by organic extraction. This was rescued by re-adding BSA-conjugated C18:0 for 2 hours, but not other fatty acids. One lipid specific to mitochondria is lipoic acid (LA). Knockdown of LA Synthase, led to reduced LA levels but not mitochondrial fragmentation, and depletion of C18:0 did not affect levels of LA or lipoylated proteins indicating the effects of C18:0 are independent of LA. Thus C18:0 regulates mitochondrial morphology in fly and human cells (Senyilmaz, 2015).
Mitochondrial fragmentation is due to either hyperactive fission or impaired fusion. Blocking fission with mdivi-1, a Drp1 inhibitor, induced mitochondrial fusion in control cells, but not in HeLas cultured without C18:0, indicating impaired mitochondrial fusion in this condition. Mitochondria labeled with photoactivatable mitoGFP rapidly fused with the rest of the network in control cells, but not in cells cultured without C18:0, demonstrating impaired fusion (Senyilmaz, 2015).
Mitochondrial fusion is regulated largely via mitofusins (Mfn). Epistasis experiments indicated C18:0 acts upstream of mitofusin to regulate mitochondrial morphology: Expression of dMfn/Marf rescued mitochondrial fragmentation in dElovl6 knockdown cells, indicating dMfn acts downstream of C18:0. C18:0 did not induce fusion in the absence of dMfn, indicating C18:0 requires dMfn for its action. Likewise, expression of dMfn rescued dElovl6- larval lethality. To test if C18:0 can rescue dMfn loss-of-function dMfn knockout flies were generated. These flies phenocopy Elovl6- mutants: they have fragmented mitochondria, reduced mitochondrial respiration, and die as early-stage larvae that do not grow. Dietary supplementation with C18:0 had no effect on growth or viability of dMfn knockouts (Senyilmaz, 2015).
It was asked if C18:0 affects Mfn via post-translational modifications (PTMs). Mfn from dElovl6- mutant larvae, or from HeLas growing with delipidated serum, migrated differently in SDS-PAGE compared to control conditions. Immunoprecipitating Mfn2 from HeLa +/-C18:0 and probing with antibodies detecting various PTMs revealed that Mfn2 from cells without C18:0 is hyper-ubiquitinated. Several ubiquitin ligases target Mfn2. Only knockdown of HUWE1 rescued the mitochondrial fragmentation and Mfn2 hyper-ubiquitination caused by C18:0 removal, as well as lethality of Elovl6 mutant flies, identifying HUWE1 as the C18:0-responsive ubiquitin ligase. As expected, increased Mfn2 ubiquitination caused Mfn2 protein destabilization. C18:0 removal did not dramatically drop Mfn2 steady-state levels, partly due to compensatory increases in Mfn2 expression, suggesting ubiquitination additionally blocks Mfn2 function in a degradation-independent manner, as for other HUWE1 targets. HUWE1 only ubiquitinates Mfn2 phosphorylated on Ser27 by JNK. Inhibition of JNK prevented mitochondrial fragmentation upon C18:0 removal. In sum, C18:0 regulates Mfn ubiquitination via HUWE1, and thereby mitochondrial morphology and function. Elovl6- mutant flies display other dMfn loss-of-function phenotypes, such as reduced ER-to-mitochondrial connections and abnormal cristae (Senyilmaz, 2015).
It was asked how C18:0 affects JNK or HUWE1 activity. ER stress can activate JNK. However C18:0 removal did not lead to a UPR response and neither knockdown of UPR effectors nor treatment with TUDCA, an ER chaperone that inhibits ER stress, blunted mitochondrial fragmentation upon C18:0 removal. Instead, it is hypothesized that C18:0 might regulate proteins via covalent binding ('stearoylation'), analogous to protein palmitoylation. C18:0 derivatives were synthesized with azide or alkyne functionalities, allowing covalent coupling to beads via copper-catalyzed azide-alkyne cycloaddition ('click chemistry'). Mltiple derivatives were tested, and only C17:0-azide induced mitochondrial fusion like C18:0. HeLas were treated with C17:0-azide for 2 hours, lysed in 8M urea to denature proteins, precipitated the lipid by coupling to beads, and covalently bound proteins were identified by mass spectrometry. The most abundant protein in the lipid pulldown was Transferrin Receptor (TfR1), confirmed by immunoblotting . Binding of TfR1 to C17:0-azide was abolished by treating lysates with hydroxylamine pH7.5, indicating a thioester linkage. The top 10 putatively stearoylated protein complexes were tested for effects on mitochondrial morphology by knockdown. Knockdown of TfR1 completely blunted mitochondrial fragmentation upon C18:0 removal. Thus TfR1 is required for cells to sense the absence of C18:0 (Senyilmaz, 2015).
Since TfR1 is important for cellular iron uptake, TfR1 stearoylation could affect mitochondria via iron uptake or delivery. However, cells grow for days in medium lacking C18:0 but die in medium lacking iron, suggesting iron uptake is not significantly impaired in the absence of C18:0. Indeed, cells in medium lacking C18:0 do not show an iron deficiency transcriptional response (ED Fig. 8b), nor a drop in protein or activity levels of enzymes containing iron-sulfur clusters, nor impaired transferrin uptake nor reduced association of transferrin with mitochondria, suggesting the effects of C18:0 are independent of iron. TfR1 also has a signaling function, activating JNK in response to the ligand gambogic acid (GA). Low concentrations of GA which do not induce apoptosis, induced rapid mitochondrial fragmentation in HeLas. This was suppressed by adding C18:0, indicating C18:0 blocks this signaling function of TfR1. Indeed, treatment of HeLas with C18:0 reduced JNK activation, using Jun kinase phosphorylation and phospho-JNK nuclear translocation as readouts. JNK inhibition blocked the ability of GA to induce mitochondrial fragmentation. In sum, these data suggest TfR1 induces mitochondrial fragmentation via JNK, and this is inhibited by TfR1 stearoylation (Senyilmaz, 2015).
It was noticed that elevating C18:0 levels in control cells increases mitochondrial fusion. Supplementing the diet of wildtype flies with C18:0 also increased mitochondrial fusion, whereas starvation of larvae led to mitochondrial fragmentation. Thus fly cells respond to both increases and decreases in levels of C18:0 (Senyilmaz, 2015).
It was asked if dietary C18:0 supplementation improves mitochondrial function in pathological conditions. Flies mutant for Pink or Parkin are established Parkinsons models. They have impaired mitochondrial function, and recapitulate Parkinsons phenotypes (reduced lifespan, neurodegeneration, and impaired motor control). Dietary supplementation with C18:0 rescued the longevity, ATP levels, and climbing defects of Pink- flies and the longevity of Parkin- flies (Senyilmaz, 2015).
Thia study has identify C18:0 as a regulator of mitochondrial function. Upon loss of C18:0, TfR1 de-stearoylation activates JNK, leading to HUWE1-dependent Mfn ubiquitination, impaired Mfn activity, and mitochondrial fragmentation. Loss of C18:0 in flies specifically impacts mitochondrial function, since Elovl6- lethality can be rescued by PGC1a, AOX, or mitofusin expression or HUWE1 knockdown. This is the first time stearoylation of a human protein regulates its function. The link of TfR1 to mitochondria perhaps makes sense since iron enters cells via TfR1 and then mainly travels to mitochondria for iron-sulfur clusters. Flies are sensitive to dietary C18:0; increased dietary C18:0 leads to increased mitochondrial fusion in vivo. Thus the metabolite C18:0 acts as a signaling molecule linking diet to mitochondrial function. Intriguingly, dietary C18:0 can improve mitochondrial function also in some pathological conditions in the fly, since dietary supplementation with C18:0 improved the Parkinsons-related phenotypes observed in Pink and Parkin mutant flies (Senyilmaz, 2015).
As fundamental processes in mitochondrial dynamics, mitochondrial fusion, fission and transport are regulated by several core components, including Miro. As an atypical Rho-like small GTPase with high molecular mass, the exchange of GDP/GTP in Miro may require assistance from a guanine nucleotide exchange factor (GEF). However, the GEF for Miro has not been identified. While studying mitochondrial morphology in Drosophila, it was incidentally observed that the loss of vimar, a gene encoding an atypical GEF, enhanced mitochondrial fission under normal physiological conditions. Because Vimar could co-immunoprecipitate with Miro in vitro, it was speculated that Vimar might be the GEF of Miro. In support of this hypothesis, a loss-of-function (LOF) vimar mutant rescued mitochondrial enlargement induced by a gain-of-function (GOF) Miro transgene; whereas a GOF vimar transgene enhanced Miro function. In addition, vimar lost its effect under the expression of a constitutively GTP-bound or GDP-bound Miro mutant background. These results indicate a genetic dependence of vimar on Miro. Moreover, mitochondrial fission was found to play a functional role in high-calcium induced necrosis, and a LOF vimar mutant rescued the mitochondrial fission defect and cell death. This result can also be explained by vimar's function through Miro, because Miro's effect on mitochondrial morphology is altered upon binding with calcium. In addition, a PINK1 mutant, which induced mitochondrial enlargement and had been considered as a Drosophila model of Parkinson's disease (PD), caused fly muscle defects, and the loss of vimar could rescue these defects. Furthermore, it was found that the mammalian homolog of Vimar, RAP1GDS1, played a similar role in regulating mitochondrial morphology, suggesting a functional conservation of this GEF member. The Miro/Vimar complex may be a promising drug target for diseases in which mitochondrial fission and fusion are dysfunctional (Ding, 2016).
Mitochondrial fission, fusion and transport play important roles for the function of this organelle. The balance between fusion and fission controls mitochondrial morphology, which is mediated by series of large dynamin-related GTPases. Among these GTPases, mitofusin1/mitofusin2 (MFN1/MFN2) and optic atrophy protein1 (OPA1) are the core components that are responsible for mitochondrial fusion, whereas dynamin-related protein 1 (Drp1) is the core component that is responsible for mitochondrial fission. In addition to these GTPases in dynamin-related family, mitochondrial Rho (Miro), an atypical member of the Rho small GTPase family, has a well-known function of transporting the mitochondria along microtubules. Miro also regulates mitochondrial morphology via inhibition of fission under physiological Ca2+ conditions, although the mechanism is not that clear. Large GTPases such as dynamin-like GTPase family members hydrolyze GTP and exchange GTP and GDP without the assistance from other regulators. However, members of the small GTPase family often require other proteins to help release their tightly bound GDP or enhance their low GTPase activities. These proteins are referred to as guanine nucleotide exchange factors (GEFs) and GTPase activating proteins (GAPs), respectively. To date, most small GTPases require unique GEFs or GAPs (Ding, 2016).
An understanding of the regulation of mitochondrial dynamics may help address many human diseases. For instance, mutations in OPA1 or MFN2 result in dominant optic atrophy or Charcot-Marie-Tooth neuropathy type 2A. Abnormal mitochondrial fission also promotes aging and cell death. In necroptosis, the formation of the necrosome promotes mitochondrial fission through dephosphorylation of Drp1. In neuronal excitotoxicity, calcium ions are overloaded, resulting in reduced levels of the MFN2 protein, which enhances mitochondrial fission and leads to neuronal necrosis In addition, other components such as Miro may participate in this process. Miro has two EF hand motifs that bind calcium; thus, Miro can couple calcium increase with reduced mitochondrial motility to meet the locally increased energy demands. Interestingly, Miro also promotes fission in the presence of excess calcium, which is distinct from its inhibitory role in fission under normal calcium concentrations. It is unclear whether Miro plays a functional role in neuronal necrosis (Ding, 2016).
The mitochondrial morphology represents a transient balance between mitochondrial fusion and fission. Using a systematic genetic screen in yeast covering approximately 88% of genes, 117 genes that regulate mitochondrial morphology were identified. Similarly, a screen of 719 genes that are predicted to encode mitochondrial proteins in worms demonstrated that more than 80% of these genes regulate mitochondrial morphology. Although many genes may regulate mitochondrial morphology, their relationships to the core mitochondrial fusion and fission components are unclear (Ding, 2016).
In studying mitochondrial morphology, it was accidently discovered that the loss of vimar (visceral mesodermal armadillo-repeats), which encodes an atypical GEF, promoted mitochondrial fission in Drosophila flight muscle cells. Furthermore, it was found that vimar was capable of interacting with Miro in vitro. Genetically, vimar required normal GDP- or GTP-bound activity of Miro to affect mitochondrial morphology, suggesting vimar is likely the Miro GEF. In addition, it was found that the Miro/Vimar complex suppressed mitochondrial fission during necrosis and mitochondrial fusion in PINK1 mutant model of Parkinson's disease (PD), making vimar a potential drug target (Ding, 2016).
Mitochondrial function can be assessed by the enzymatic activity of citrate synthase (CS), the first enzyme in the Krebs cycle that converts acetyl-CoA and oxaloacetate to citrate. In cultured Drosophila S2 cells, vimar knock down by RNAi resulted in reduced CS activity, indicating that vimar may positively regulate mitochondrial function. Because mitochondrial fission has generally been associated with reduced mitochondrial respiration, the decreased CS activity may be a result of mitochondrial fission. Consistent with this notion, the results demonstrated that the LOF of vimar promoted mitochondrial fission. In addition, a GOF vimar transgene had a minimal effect on mitochondrial morphology, indicating that vimar activity might be saturated under normal physiological conditions (Ding, 2016).
Because Vimar has been predicted to be a GEF, it was hypothesized that Vimar may regulate mitochondrial morphology by affecting a small GTPase, which requires a GEF to help with the GTP/GDP exchange process. Interestingly, Miro is one such small GTPase that is known to play important roles in mitochondrial fission and transport. It is proposed that Vimar and Miro may function as a complex. First, a fraction of the Vimar protein was localized to the mitochondria, possibly indicating a functional role on mitochondria. Interestingly, the mitochondrial localization of Vimar seems not dependent on Miro, because LOF Miro did not affect the mitochondrial fraction of Vimar. This indicates that Vimar may directly bind with mitochondria or through other scaffolding proteins. Second, Vimar and Miro could physically interact with each other, at least in vitro. Their interaction seems not affected by the GTPase activity of Miro, because the constitutively GDP- or GTP-bound Miro mutants did not affect their interactions. Third, vimar genetically interacted with Miro. This included the result demonstrating that the LOF vimar mutant reduced the effect of Miro on mitochondrial fission inhibition and the GOF vimar transgene had the opposite effect. Moreover, in the constitutive GFP-bound or GDP-bound Miro mutants, the effect of the GOF or LOF vimar was abolished. Therefore, Vimar requires the normal GDP/GTP binding activity of Miro to function. It is also known that Miro1 overexpression increase mitochondrial size partially by suppression of the Drp1 function. Consistently, increased mitochondrial fission in the LOF of Miro or vimar was abolished by loss of Drp1, suggesting the Miro/vimar complex depends on Drp1 to regulate mitochondrial morphology (Ding, 2016).
Familial PD caused by mutations in PINK1 or Parkin results in a series of mitochondrial dysfunctions, particularly the failure to eliminate damaged mitochondria through mitophagy. In these PINK1 or Parkin mutants, the key proteins involved in mitochondrial fusion and fission, such as Marf/Mitofusin and Miro, accumulate. In the PINK1 mutant flies, the flight muscle is damaged, resulting in wing posture defects. Similarly, it was observed that Miro overexpression in the flight muscle resulted in a strong wing posture defect. This result may explain the wing posture defect in the PINK1 mutant, in which the levels of the Miro protein are increased. This study demonstrated that the LOF of vimar could rescue the wing defect in the PINK1 mutant, consistent with the hypothesis that vimar functions through Miro (Ding, 2016).
When the intracellular calcium level is high, Miro switches from promoting mitochondrial fission inhibition to enhancing mitochondrial fission. The mechanism for this switch is unclear, although alterations of Drp1 function could be one possibility. Interestingly, Gem1, the yeast homolog of Miro GTPase, has been reported to function as a negative regulator for ER-mitochondria contacts, where Drp1 aggregates and cleaves mitochondria into smaller units. This may serve as the mechanism for Miro to regulate mitochondrial morphology via Drp1. In addition to affect mitochondrial fission, Miro also regulates mitochondrial transport in a calcium dependent manner. For mitochondrial transport, Miro forms protein complexes with Milton, a kinesin adaptor, and with motor proteins, such as kinesin and dynein. In high calcium conditions, Miro alters its binding patterns and results in reduced transport activity. Based on these reports, it is proposed that the Miro/Vimar complex acts together to affect mitochondrial morphology: at normal condition, Miro/Vimar inhibits fission via Drp1; at high calcium state, Ca2+ bound Miro switches its function to promote fission. Indeed, Vimar responds to the calcium change in the same way as Miro. In addition, the data demonstrated that knocking down RAP1GDS1 and Miro1 increased mitochondrial fission and could rescue calcium overload induced necrosis, similar to the loss of Vimar or Miro in Drosophila. These data support the hypothesis that RAP1GDS1 is the mammalian homolog of Vimar, supporting a previous prediction (Ding, 2016).
Mitochondrial fission plays important role in apoptosis by promoting mitochondrial outer-membrane permeabilization (MOMP) to release cytochrome c from the mitochondria. The use of the Drp1 inhibitor mdivi to block fission has been shown to be an effective treatment for stroke, and the function of mitochondrial fission on necrotic cell death has been well documented. The uncertainty lies in the lack of genetic evidence and downstream mechanism of mitochondrial fission in necrosis. The current data demonstrated that mitochondrial fragmentation occurs in necrotic neurons, and the LOF Drp1 and vimar mutants both suppressed neuronal necrosis (Ding, 2016).
Much evidence suggests that the mitochondrial fusion and fission defects are directly linked to many human diseases, and strategies that target the Miro/vimar complex may affect a broad spectrum of diseases. For instance, mutations in the fragile X mental retardation 1 (FMR1) gene, which result from expansion of trinucleotide repeat in the 5' untranslated region, often cause enhanced mitochondrial fission and mental retardation syndrome. Likewise, aberrant mitochondrial fusion was observed in a Drosophila Alzheimer's disease model induced by the ectopic expression of a human tau mutant (tauR406W). In this case, the tau mutant may promote excessive actin stabilization to decrease Drp1 recruitment to the mitochondria, which results in excessive mitochondrial fusion and neurodegeneration. Due to the dual function of the Miro/Vimar complex in high-Ca2+ induced necrosis and PINK1 mutant induced PD, a drug to target this complex may benefit both disease states. As a modulator, it may be safer to target Vimar/ RAP1GDS1 (Ding, 2016).
In eukaryotic cells, mitochondria form a dynamic interconnected network to respond to changing needs at different subcellular locations. A fundamental yet unanswered question regarding this network is whether, and if so how, local fusion and fission of individual mitochondria affect their global distribution. To address this question, high-resolution computational image analysis techniques have been developed to examine the relations between mitochondrial fusion/fission and spatial distribution within the axon of Drosophila larval neurons. Stationary and moving mitochondria were found to undergo fusion and fission regularly but followed different spatial distribution patterns and exhibited different morphology. Disruption of inner membrane fusion by knockdown of dOpa1, Drosophila Optic Atrophy 1, not only increased the spatial density of stationary and moving mitochondria but also changed their spatial distributions and morphology differentially. Knockdown of dOpa1 also impaired axonal transport of mitochondria. But the changed spatial distributions of mitochondria resulted primarily from disruption of inner membrane fusion because knockdown of Milton, a mitochondrial kinesin-1 adapter, caused similar transport velocity impairment but different spatial distributions. Together, these data reveals that stationary mitochondria within the axon interconnect with moving mitochondria through fusion and fission and that local inner membrane fusion between individual mitochondria mediates their global distribution (Yu, 2016).
Although individual mitochondria within the axon may appear as discrete compartments, they interconnect through fusion and fission as well as transport and anchoring to form a dynamic network. This network is distributed spatially to fulfill changing needs at different locations. Proper spatial distribution of axonal mitochondria has been shown, for example, to be essential for axon branching, synaptic functions and a variety of other neuronal activities. This study has focused on addressing a basic question regarding the axonal mitochondrial network, namely whether, and if so how, local fusion and fission of individual mitochondria affect their global distribution (Yu, 2016).
Inner membrane fusion, which occurred locally between individual mitochondria, mediates the global distribution of mitochondria within the axon. In wild-type Drosophila larvae, spatial distribution of stationary and moving axonal mitochondria followed distinct patterns. Disruption of inner membrane fusion by dOpa1 knockdown not only caused dramatic imbalance between fusion and fission and fragmentation of individual mitochondria but also changed their spatial distribution patterns, resulting in progressive loss of both stationary and moving mitochondria along the axon towards distal axon regions. Consistent with this result, disruption of mitochondrial outer membrane fusion by knocking down Marf (Drosophila ortholog Mfn2) resulted in similar progressive loss of stationary and moving mitochondria along the axon. On the other hand, knockdown of mitochondrial motor adaptor Milton impaired retrograde velocities of mitochondrial transport similarly as dOpa1 knockdown but changed the spatial distribution of axonal mitochondria differently. Together, these results showed that the changes to the spatial distributions of axonal mitochondria under dOpa1 knockdown were caused primarily by disruption of inner membrane fusion and that impairment of mitochondrial transport under dOpa1 knockdown played a secondary role in causing these changes. Therefore, local inner membrane fusion plays an important role in mediating the global spatial distribution of mitochondria (Yu, 2016).
This study is in agreement with a growing number of studies suggesting that, in addition to mediating local mitochondrial content exchange or transport-docking, fusion and fission are involved in regulating the global organization of the mitochondrial network, although direct analysis of the relations between local fusion/fission and the global organization of the mitochondrial network was lacking in previous studies. The current data reveals direct connections and quantitative relations between mitochondrial inner membrane fusion and spatial distribution. However, a limitation of this assay is that it is restricted to the specific group of neurons in which SG26 Gal4 is expressed. Further studies should examine relations between mitochondrial fusion/fission and spatial distribution in different groups of neurons (Yu, 2016).
A unique challenge facing neurons is to sustain functionally competent mitochondria over extended distances. Indeed, neurons are known to be particularly vulnerable to dysfunction of mitochondria and mutations of mitochondrial proteins. Since the soma of neurons is the primary site for biogenesis and degradation of mitochondria, a basic question regarding axonal mitochondria is how they stay functionally competent and renew themselves while being far away from the neuronal cell body. The current data supports the hypothesis that axonal mitochondria replenish themselves through fusion with moving ones passing by and through fission to discard their damaged portion. Specifically, it was found that within the axon of Drosophila larval neurons, stationary mitochondria underwent fusion and fission regularly with moving mitochondria. They were also much larger than moving ones but became more fragmented under dOpa1 knockdown. Together, the data suggests differential roles of stationary and moving mitochondria: while stationary mitochondria fulfill metabolic and functional needs of their local areas, moving mitochondria support stationary mitochondria by renewing their content through fusion/fission. Furthermore, moving mitochondria can move to areas where new needs arise and settle down as stationary mitochondria. The data supports the quality control model of axonal mitochondria previously proposed47 but does not rule out the possibility that populations of stationary and moving mitochondria interchange through direct switching between their motion states by a transport-docking mechanism (Yu, 2016).
Based on the data, a model is proposed of how the spatial distribution of axonal mitochondria is maintained in healthy neurons and how it is changed by disruption of inner membrane fusion. It is conjectured that anchored stationary mitochondria activate a fusion signal when renewal is needed. The fusion signal retains some of the passing mitochondria to engage in fusion and fission. Successful completion of fusion and fission inactivates the signal, allowing moving mitochondria to simply pass by. When inner membrane fusion is disrupted by OPA1 knockdown, the fusion signal of stationary mitochondria can no longer be inactivated because fusion cannot be completed. Cristae disorganization under dOpa1 knockdown may also interfere with the successful fusion of mitochondria. The stationary mitochondria close to the soma and with activated fusion signal will retain increasing numbers of moving mitochondria so that fewer moving mitochondria can reach more distal regions. This results in a gradual accumulation of stationary mitochondria close to the soma and progressive loss of mitochondria along the axon towards distal synaptic terminals. The loss of mitochondria in distal regions eventually leads to neurodegeneration (Yu, 2016).
Mitochondria contain their own genomes that, unlike nuclear genomes, are inherited only in the maternal line. Owing to a high mutation rate and low levels of recombination of mitrochondrial DNA (mtDNA), special selection mechanisms exist in the female germline to prevent the accumulation of deleterious mutations. However, the molecular mechanisms that underpin selection are poorly understood. This study visualized germline selection in Drosophila using an allele-specific fluorescent in situ-hybridization approach to distinguish wild-type from mutant mtDNA. Selection first manifests in the early stages of Drosophila oogenesis, triggered by reduction of the pro-fusion protein Mitofusin. This leads to the physical separation of mitochondrial genomes into different mitochondrial fragments, which prevents the mixing of genomes and their products and thereby reduces complementation. Once fragmented, mitochondria that contain mutant genomes are less able to produce ATP, which marks them for selection through a process that requires the mitophagy proteins Atg1 and BNIP3. A reduction in Atg1 or BNIP3 decreases the amount of wild-type mtDNA, which suggests a link between mitochondrial turnover and mtDNA replication. Fragmentation is not only necessary for selection in germline tissues, but is also sufficient to induce selection in somatic tissues in which selection is normally absent. It is postulated that there is a generalizable mechanism for selection against deleterious mtDNA mutations, which may enable the development of strategies for the treatment of mtDNA disorders (Lieber, 2019).
The function of AarF domain-containing kinase 1 (ADCK1) has not been thoroughly revealed. This study identified that ADCK1 utilizes YME1-like 1 ATPase (YME1L1) to control optic atrophy 1 (OPA1) and inner membrane mitochondrial protein (IMMT) in regulating mitochondrial dynamics and cristae structure. It was firstly observed that a serious developmental impairment occurred in Drosophila ADCK1 (dADCK1) deletion mutant, resulting in premature death before adulthood. By using temperature sensitive ubiquitously expression driver tub-Gal80ts/tub-Gal4 or muscle-specific expression driver mhc-Gal4, severely defective locomotive activities and structural abnormality in the muscle were observed along with increased mitochondrial fusion in the dADCK1 knockdown flies. Moreover, decreased mitochondrial membrane potential, ATP production and survival rate along with increased ROS and apoptosis in the flies further demonstrated that the structural abnormalities of mitochondria induced by dADCK1 knockdown led to their functional abnormalities. Consistent with the ADCK1 loss-of-function data in Drosophila, ADCK1 over-expression induced mitochondrial fission and clustering in addition to destruction of the cristae structure in Drosophila and mammalian cells. Interestingly, knockdown of YME1L1 rescued the phenotypes of ADCK1 over-expression. Furthermore, genetic epistasis from fly genetics and mammalian cell biology experiments led to a discovery the interactions among IMMT, OPA1 and ADCK1. Collectively, these results established a mitochondrial signaling pathway composed of ADCK1, YME1L1, OPA1 and IMMT, which has essential roles in maintaining mitochondrial morphologies and functions in the muscle (Yoon, 2019).
The enzyme kynurenine 3-monooxygenase (KMO) operates at a critical branch-point in the kynurenine pathway (KP), the major route of tryptophan metabolism. As the KP has been implicated in the pathogenesis of several human diseases, KMO and other enzymes that control metabolic flux through the pathway are potential therapeutic targets for these disorders. While KMO is localized to the outer mitochondrial membrane in eukaryotic organisms, no mitochondrial role for KMO has been described. In this study, KMO (cinnabar) deficient Drosophila melanogaster were investigated for mitochondrial phenotypes in vitro and in vivo. A loss of function allele or RNAi knockdown of the Drosophila KMO ortholog (cinnabar) causes a range of morphological and functional alterations to mitochondria, which are independent of changes to levels of KP metabolites. Notably, cinnabar genetically interacts with the Parkinson's disease associated genes Pink1 and parkin, as well as the mitochondrial fission gene Drp1, implicating KMO in mitochondrial dynamics and mitophagy, mechanisms which govern the maintenance of a healthy mitochondrial network. Overexpression of human KMO in mammalian cells finds that KMO plays a role in the post-translational regulation of DRP1. These findings reveal a novel mitochondrial role for KMO, independent from its enzymatic role in the kynurenine pathway (Maddison, 2020).
Balanced mitochondrial fission and fusion play an important role in shaping and distributing mitochondria, as well as contributing to mitochondrial homeostasis and adaptation to stress. In particular, mitochondrial fission is required to facilitate degradation of damaged or dysfunctional units via mitophagy. Two Parkinson's disease factors, PINK1 and Parkin, are considered key mediators of damage-induced mitophagy, and promoting mitochondrial fission is sufficient to suppress the pathological phenotypes in Drosophila Pink1/parkin mutants. Additional factors were sought that impinge on mitochondrial dynamics and which may also suppress Pink1/parkin phenotypes. The Drosophila phosphatidylinositol 4-kinase IIIβ homologue, Four wheel drive (Fwd), promotes mitochondrial fission downstream of the pro-fission factor Drp1. Previously described only as male sterile, this study identified several new phenotypes in fwd mutants, including locomotor deficits and shortened lifespan, which are accompanied by mitochondrial dysfunction. Finally, fwd overexpression can suppress locomotor deficits and mitochondrial disruption in Pink1/parkin mutants, consistent with its function in promoting mitochondrial fission. Together these results shed light on the complex mechanisms of mitochondrial fission and further underscore the potential of modulating mitochondrial fission/fusion dynamics in the context of neurodegeneration (Terriente-Felix, 2020).
The fate and proliferative capacity of stem cells have been shown to strongly depend on their metabolic state. Mitochondria are the powerhouses of the cell being responsible for energy production via oxidative phosphorylation (OxPhos) as well as for several other metabolic pathways. Mitochondrial activity strongly depends on their structural organization, with their size and shape being regulated by mitochondrial fusion and fission, a process known as mitochondrial dynamics. However, the significance of mitochondrial dynamics in the regulation of stem cell metabolism and fate remains elusive. This study characterized the role of mitochondria morphology in female germ stem cells (GSCs) and in their more differentiated lineage. Mitochondria are particularly important in the female GSC lineage. Not only do they provide these cells with their energy requirements to generate the oocyte but they are also the only mitochondria pool to be inherited by the offspring. The undifferentiated GSCs predominantly have fissed mitochondria, whereas more differentiated germ cells have more fused mitochondria. By reducing the levels of mitochondrial dynamics regulators, it was shown that both fused and fissed mitochondria are required for the maintenance of a stable GSC pool. Surprisingly, it was found that disrupting mitochondrial dynamics in the germline also strongly affects nurse cells morphology, impairing egg chamber development and female fertility. Interestingly, reducing the levels of key enzymes in the Tricarboxylic Acid Cycle (TCA), known to cause OxPhos reduction, also affects GSC number. This defect in GSC self-renewal capacity indicates that at least basal levels of TCA/OxPhos are required in GSCs. These findings show that mitochondrial dynamics is essential for female GSC maintenance and female fertility, and that mitochondria fusion and fission events are dynamically regulated during GSC differentiation, possibly to modulate their metabolic profile (Garcez, 2020).
Mitochondria are highly dynamic organelles with strict quality control processes that maintain cellular homeostasis. Within axons, coordinated cycles of fission-fusion mediated by dynamin related GTPase protein (DRP1) and mitofusins (MFN), together with regulated motility of healthy mitochondria anterogradely and damaged/oxidized mitochondria retrogradely, control mitochondrial shape, distribution and size.
This study has isolated the mechanistic role of α-syn in mitochondrial homeostasis in vivo in a humanized Drosophila model of Parkinson's disease (PD). It was shown that excess α-syn causes fragmented mitochondria, which persists with either truncation of the C-terminus (α(1-120)) or deletion of the NAC region (α(ΔNAC)). Using in vivo oxidation reporters Mito-roGFP2-ORP1/GRX1 and MitoTimer, it was found that α-mediated fragments were oxidized/damaged, but α(1-120)-induced fragments were healthy, suggesting that the C-terminus is required for oxidation. α-mediated oxidized fragments showed biased retrograde motility, but α(1-120)-mediated healthy fragments did not, demonstrating that the C-terminus likely mediates the retrograde motility of oxidized mitochondria. Depletion/inhibition or excess DRP1-rescued α-syn-mediated fragmentation, oxidation, and the biased retrograde motility, indicating that DRP1-mediated fragmentation is likely upstream of oxidation and motility changes. Further, excess PINK/Parkin, two PD-associated proteins that function to coordinate mitochondrial turnover via induction of selective mitophagy, rescued α-syn-mediated membrane depolarization, oxidation and cell death in a C-terminus-dependent manner, suggesting a functional interaction between &alpha-syn and PINK/Parkin. Taken together, these findings identify distinct roles for α-syn in mitochondrial homeostasis, highlighting a previously unknown pathogenic pathway for the initiation of PD (Krzystek, 2021).
The echanism by which Mitochondria morphology and dynamics regulate cell differentiation and lineage choice remains incompletely understood. Asrij/OCIAD1 is a conserved protein that governs mitochondrial morphology, energy metabolism and human embryonic stem cell (hESC) differentiation. This study compared hESC phenotypes with those of Drosophila hematopoiesis, where Asrij is shown to regulate blood progenitor maintenance by conserved mechanisms. In concordance with hESC studies, this study found that Drosophila Asrij also localizes to mitochondria of larval blood cells and its depletion from progenitors results in elongated mitochondria. Live imaging of asrij knockdown hemocytes and of OCIAD1 knockout hESCs showed reduced mitochondrial dynamics. It was hypothesized that mitochondrial fission and fusion may control progenitor maintenance or differentiation in an Asrij-dependent manner. Knockdown of the fission regulator Drp1 in Drosophila lymph gland progenitors specifically suppressed crystal cell differentiation whereas depletion of the fusion regulator Marf (Drosophila Mitofusin) increased the same with concomitant upregulation of Notch signaling. These phenotypes were stronger in anterior progenitors and were exacerbated by Asrij depletion. Asrij is known to suppress Notch signaling and crystal cell differentiation. This study demonstrates a conserved role for Asrij/OCIAD1 in linking mitochondrial dynamics and progenitor differentiation (Ray, 2021).
A healthy population of mitochondria, maintained by proper fission, fusion, and degradation, is critical for the long-term survival and function of neurons. In this study, the discovery of mitophagy intermediates in fission-impaired Drosophila neurons brings new perspective into the relationship between mitochondrial fission and mitophagy. Neurons lacking either the ataxia disease gene Vps13D or the dynamin related protein Drp1 contain enlarged mitochondria that are engaged with autophagy machinery and also lack matrix components. Reporter assays combined with genetic studies imply that mitophagy both initiates and is completed in Drp1 impaired neurons, but fails to complete in Vps13D impaired neurons, which accumulate compromised mitochondria within stalled mito-phagophores. These findings imply that in fission-defective neurons, mitophagy becomes induced, and that the lipid channel containing protein Vps13D has separable functions in mitochondrial fission and phagophore elongation (Insolera, 2021).
Optimal mitochondrial function determined by mitochondrial dynamics, morphology and activity is coupled to stem cell differentiation and organism development. However, the mechanisms of interaction of signaling pathways with mitochondrial morphology and activity are not completely understood. This study assessed the role of mitochondrial fusion and fission in the differentiation of neural stem cells called neuroblasts (NB) in the Drosophila brain. Depleting mitochondrial inner membrane fusion protein Opa1 and mitochondrial outer membrane fusion protein Marf in the Drosophila type II NB lineage led to mitochondrial fragmentation and loss of activity. Opa1 and Marf depletion did not affect the numbers of type II NBs but led to a decrease in differentiated progeny. Opa1 depletion decreased the mature intermediate precursor cells (INPs), ganglion mother cells (GMCs) and neurons by the decreased proliferation of the type II NBs and mature INPs. Marf depletion led to a decrease in neurons by a depletion of proliferation of GMCs. On the contrary, loss of mitochondrial fission protein Drp1 led to mitochondrial clustering but did not show defects in differentiation. Depletion of Drp1 along with Opa1 or Marf also led to mitochondrial clustering and suppressed the loss of mitochondrial activity and defects in proliferation and differentiation in the type II NB lineage. Opa1 depletion led to decreased Notch signaling in the type II NB lineage. Further, Notch signaling depletion via the canonical pathway showed mitochondrial fragmentation and loss of differentiation similar to Opa1 depletion. An increase in Notch signaling showed mitochondrial clustering similar to Drp1 mutants. Further, Drp1 mutant overexpression combined with Notch depletion showed mitochondrial fusion and drove differentiation in the lineage, suggesting that fused mitochondria can influence differentiation in the type II NB lineage. These results implicate crosstalk between proliferation, Notch signaling, mitochondrial activity and fusion as an essential step in differentiation in the type II NB lineage (Dubal, 2022).
Stem cells often possess immature mitochondria with few inner membrane invaginations, which increase as stem cells differentiate. Despite this being a conserved feature across many stem cell types in numerous organisms, how and why mitochondria undergo such remodelling during stem cell differentiation has remained unclear. Using Drosophila germline stem cells (GSCs), this study shows that Complex V drives mitochondrial remodelling during the early stages of GSC differentiation, prior to terminal differentiation. This endows germline mitochondria with the capacity to generate large amounts of ATP required for later egg growth and development. Interestingly, impairing mitochondrial remodelling prior to terminal differentiation results in endoplasmic reticulum (ER) lipid bilayer stress, Protein kinase R-like ER kinase (PERK)-mediated activation of the Integrated Stress Response (ISR) and germ cell death. Taken together, these data suggest that mitochondrial remodelling is an essential and tightly integrated aspect of stem cell differentiation. This work sheds light on the potential impact of mitochondrial dysfunction on stem and germ cell function, highlighting ER lipid bilayer stress as a potential major driver of phenotypes caused by mitochondrial dysfunction (Monteiro, 2023).
Mitochondria undergo structural changes reflective of functional statuses. Ultrastructural characterizing of mitochondria is valuable for understanding mitochondrial dysfunction in various pathological conditions. PINK1, a Parkinson's disease (PD) associated gene, plays key roles in maintaining mitochondrial function and integrity. In Drosophila melanogaster, deficiency of PINK1 results in PD-like pathologies due to mitochondrial abnormalities. This study reports the existence of a new type of mitochondrial-membrane deformity, mitochondrial spherical compartmentation (MSC), caused by PINK1 deficiency in Drosophila. The MSC is a three-dimensional spheroid-like mitochondrial membrane structure encompassing nonselective contents. Upregulation of dDrp1, downregulation of dMarf, and upregulation of dArgK1-A-all resulting in mitochondrial fragmentation-were able to suppress the formation of MSC. Furthermore, arginine kinase, only when localizing to the vicinity of mitochondria, induced mitochondrial fragmentation and reversed the MSC phenotype. In summary, this study demonstrates that loss of dPINK1 leads to the formation of mitochondrial-membrane deformity MSC, which responds to mitochondrial dynamics. In addition, these data suggest a new perspective of how phosphagen energy-buffer system might regulate mitochondrial dynamics (Li, 2023).
Mitochondria located within neuronal presynaptic terminals have been shown to play important roles in the release of chemical neurotransmitters. In the present study, a genetic screen for synaptic transmission mutants of Drosophila has identified the first mutation in a Drosophila homolog of the mitochondrial protein P32. Although P32 is highly conserved and has been studied extensively, its physiological role in mitochondria remains unknown and it has not previously been implicated in neural function. The Drosophila P32 mutant, referred to as dp32EC1, exhibited a temperature-sensitive (TS) paralytic behavioral phenotype. Moreover, electrophysiological analysis at adult neuromuscular synapses revealed a TS reduction in the amplitude of excitatory postsynaptic currents (EPSC) and indicated that dP32 functions in neurotransmitter release. These studies are the first to address P32 function in Drosophila and expand the knowledge of mitochondrial proteins contributing to synaptic transmission (Lutas, 2012).
A genetic screen for synaptic transmission mutants in Drosophila isolated a new mutation in a Drosophila homolog of the mitochondrial protein P32, which represents the first P32 mutation in a multicellular organism. Although P32 is highly conserved and has been studied extensively, its physiological function in mitochondria remains unknown. This new mutant, referred to as dP32EC1, exhibited a temperature-sensitive (TS) paralytic behavioral phenotype. Moreover, electrophysiological analysis at adult neuromuscular synapses revealed a TS reduction in neurotransmitter release, indicating that dP32 serves an important function in synaptic transmission. Immunocytochemical analysis has shown that dP32 is located within presynaptic mitochondria, which are known to be important in ATP production and calcium signaling at synapses. Furthermore, the basic molecular and structural organization of synapses appears to be normal in the dP32 mutant, suggesting a direct role for this protein in synaptic function. At the molecular level, biochemical studies indicated conserved homomultimeric interactions of dP32 subunits. Finally, assessment of presynaptic mitochondrial function was examined in the dP32 mutant through measurement of ATP levels and imaging studies of mitochondrial membrane potential and presynaptic calcium. This work indicated that mitochondrial ATP production and membrane potential in the dP32 mutant resembled wild-type, whereas the mutant exhibited a TS increase in both resting and evoked presynaptic calcium concentration. Taken together, the preceding findings reveal a role for dP32 in synaptic transmission and mitochondrial regulation of presynaptic calcium (Lutas, 2012).
Mitochondrial localization of P32 proteins involves an N-terminal targeting domain that is cleaved from the mature targeted protein. Comparison of Drosophila and vertebrate P32 sequences indicates conservation of the proteolytic cleavage site in dP32. In the present study, an equivalent targeting function for the N-terminal domain of dP32 was demonstrated through its ability to mediate mitochondrial targeting. When the first 71 amino acids of dP32, including the proteolytic cleavage site, was fused to GCaMP3, this fusion protein (mito-GCaMP3) was efficiently targeted to mitochondria. Although only modest sequence conservation was observed between the N-terminal domains of dP32 and vertebrate P32 proteins, previous studies suggest that mitochondrial targeting domains vary in amino acid sequence but often share an amphipathic helical structure (Lutas, 2012).
Structural studies have established that P32 is a homotrimer in which monomers are arranged around a central pore in a donut-like structure. In the present study, homomultimerization of dP32 subunits was demonstrated in co-immunoprecipitation experiments. The trimeric structure of P32 exhibits a highly asymmetric charge distribution that creates a concentration of negatively charged residues along one side of the donut, raising the possibility that P32 may participate in calcium binding within the mitochondrial matrix. Notably, five residues that are spatially clustered to form a pocket on the negatively charged side of human P32, Glu-89, Leu-231, Asp-232, Glu-264, and Tyr-268, are identical in the Drosophila protein. Further genetic analysis may address the importance of these clustered residues in dP32 function at synapses (Lutas, 2012).
Several possible mechanisms of dP32 function in mitochondria and synaptic transmission were considered and investigated in this paper, most notably possible roles in supporting mitochondrial membrane potential, ATP production, and presynaptic calcium signaling. Among these, the observations favor a function for dP32 in mitochondrial mechanisms regulating presynaptic calcium. Although neurotransmitter release was reduced at restrictive temperatures in dP32EC1, the presynaptic calcium concentration was increased both at rest and in response to synaptic stimulation. It is of interest to consider why the increase in presynaptic calcium in dP32EC1 is TS in what appears to be a complete loss-of-function mutant. Previous studies at Drosophila larval neuromuscular synapses at elevated temperatures have observed a TS increase in resting cytosolic calcium and associated inhibition of neurotransmitter release. This calcium increase was enhanced by pharmacological inhibition of presynaptic calcium clearance mechanisms or genetic removal of presynaptic mitochondria, but it remained dependent on temperature. The present findings may reflect a similar TS process involving calcium-dependent inhibition of neurotransmitter release and dP32-dependent mitochondrial mechanisms. Efforts to further address these mechanisms were pursued by employing a calcium indicator targeted to the mitochondrial matrix, mito-GCaMP3. Although this approach was successful for imaging mitochondrial calcium transients elicited by motor axon stimulation in both WT and dP32EC1 at 20°C, robust calcium transients could not be observed in either genotype when the temperature was increased to the restrictive temperatures of 33° or 36° (Lutas, 2012).
The preceding observations suggest that sustained elevation of presynaptic calcium in the dP32 mutant may lead to reduced neurotransmitter release. Such a calcium-dependent mechanism has been reported previously in the squid giant synapse and attributed to calcium-dependent adaptation of the neurotransmitter release apparatus. Understanding the precise mechanism by which loss of dP32 impairs neurotransmitter release will require further investigation. One interesting question is how the absence of dP32 in the mitochondrial matrix leads to increased presynaptic calcium and whether this reflects the putative calcium binding capacity of this protein. Finally, while the present study is focused on the newly discovered role for P32 in neurotransmitter release, the resulting research materials are expected to facilitate in vivo analysis of P32 function in a broad range of biological processes (Lutas, 2012).
Mutations in the mitochondrial Ser/Thr kinase PINK1 cause Parkinson's disease. One of the substrates of PINK1 is the outer mitochondrial membrane protein Miro, which regulates mitochondrial transport. This study uncovered novel physiological functions of PINK1-mediated phosphorylation of Miro, using Drosophila as a model. Endogenous Drosophila Miro (DMiro) was replaced with transgenically expressed wildtype, or mutant DMiro predicted to resist PINK1-mediated phosphorylation. The expression of phospho-resistant DMiro in a DMiro null mutant background was found to phenocopy a subset of phenotypes of PINK1 null. Specifically, phospho-resistant DMiro increased mitochondrial movement and synaptic growth at larval neuromuscular junctions, and decreased the number of dopaminergic neurons in adult brains. Therefore, PINK1 may inhibit synaptic growth and protect dopaminergic neurons by phosphorylating DMiro. Furthermore, muscle degeneration, swollen mitochondria and locomotor defects found in PINK1 null flies were not observed in phospho-resistant DMiro flies. Thus, this study established an in vivo platform to define functional consequences of PINK1-mediated phosphorylation of its substrates (Tsai, 2014).
Mutations in the Ser/Thr kinase PINK1 (PTEN-induced Putative Kinase 1) cause Parkinson's disease (PD), one of the most common neurodegenerative disorders. Emerging evidence suggests that PINK1 functions upstream of another PD-associated protein, the E3 ubiquitin ligase Parkin, to clear damaged mitochondria via mitophagy. How PINK1 primes cytosolic Parkin for mitophagy remains unclear, although PINK1-mediated phosphorylation of Parkin or ubiquitin may be involved. Previous work has shown that PINK1, in cooperation with Parkin, also regulates mitochondrial trafficking by controlling turn-over of Miro (Wang, 2011), an outer mitochondrial membrane (OMM) protein that anchors the kinesin and dynein motors to mitochondria. Work in cultured cells has demonstrated that mitochondrial depolarization or damage stabilizes PINK1 on the OMM. Concomitantly, PINK1 phosphorylates Miro, which then activates proteasomal degradation of Miro in a Parkin-dependent manner and arrests mitochondrial transport. This may serve as a critical step in quarantining damaged mitochondria prior to their degradation via mitophagy. However, the physiological significance of PINK1-mediated phosphorylation of Miro in vivo has not yet been determined (Tsai, 2014).
Recent studies have shown that Mitofusin, another OMM protein, is also a common substrate for both PINK1 and Parkin. Mitofusin facilitates mitochondrial fusion, and mitochondrial damage rapidly degrades Mitofusion causing mitochondria to fragment prior to mitophagy. PINK1 also phosphorylates the anti-apoptotic protein Bcl-xL on the OMM of depolarized mitochondria, not to regulate mitophagy, but to prevent cell death. In addition to the PINK1 OMM substrates Miro, Mitofusin and Bcl-xL, PINK1 mediates phosphorylation of the mitochondrial chaperon TRAP1 and the Serine protease HtrA2, which are both located in the mitochondrial inter-membrane space. This wide range of the potential substrates of PINK1 suggests that it may have multiple cellular functions (Tsai, 2014).
The consensus target sequence for phosphorylation by PINK1 has remained elusive. To date, Miro is the only PINK1 mitochondrial substrate whose phosphorylation residues have been determined (b4 (Wang, 2011). Two Drosophila Miro (DMiro) peptides with a high degree of similarity to the human sequence were identified as potential targets of PINK1-mediated phosphorylation in vitro. This study has determined the critical role of PINK1 phosphorylation sites on DMiro for maintaining neuronal homeostasis and protecting dopaminergic (DA) neurons in vivo. Drosophila is a robust genetic and cellular tool for modeling human neurodegenerative diseases. Loss of PINK1 in Drosophila mimics many aspects of PD pathology, including a severe loss of DA neurons, which is a hallmark of PD. However, few of the molecular and cellular mechanisms underlying the behavioral and cellular phenotypes of PINK1 null mutant flies have been clearly defined. This study identifies that DMiroS182A,S324A,T325A, which is predicted to resist PINK1-mediated phosphorylation, causes increased mitochondrial movement, synaptic overgrowth, and loss of DA neurons. All three of these defects are also observed in PINK1 null mutant flies. Hence, this work suggests that Miro is a crucial substrate for causing these phenotypes by mutant PINK1. This study opens a new door to fully dissect PINK1 functions by studying its individual substrates. Since PINK1-related hereditary PD shares symptomatic and pathological similarities with the majority of idiopathic PD, such work will advance understanding of the cellular and molecular underpinnings of PD's destructive path (Tsai, 2014).
Extensive studies using cell cultures have established a critical role for PINK1 in damage-induced mitophagy. PINK1/Parkin-dependent regulation of mitochondrial transport by controlling Miro protein levels on mitochondria is likely a key step prior to initiating mitophagy in cultured neurons. This study show that PINK1-mediated phosphorylation of DMiro is required for normal mitochondrial movement in axon terminals, synaptic growth, and the neuroprotection of DA neurons. Importantly, loss of PINK1-mediated phosphorylation of DMiro has no significant effect on the mitochondrial membrane potential, excluding the possibility that the observed phenotypic effects are due to an impairment of mitophagy and an accumulation of damaged mitochondria. Accordingly, under these conditions PINK1-mediated phosphorylation of DMiro may not be required for mitophagy. However, this does not necessarily contradict its mitophagic role; rather, this represents circumstances under which its mitophagic role is dispensable. It is tempting to speculate that an efficient regulation of mitophagy is more critical in aging neurons (Tsai, 2014).
These studies identify a conserved site in human and Drosophila Miro, MiroSer156/DMiroSer182, to be a main residue for PINK1-mediated phosphorylation. Additional conserved sites were found in DMiro that may have a cooperative role. Future studies determining their functions in mammalian systems are warranted to confirm if a similar regulatory mechanism is at play. This study suggests that these PINK1 phosphorylation sites in DMiro are not absolutely required for the subsequent Parkin-dependent degradation of DMiro, because when harsh treatment of mitochondrial uncoupler CCCP is applied, the phospho-resistant DMiroS182A,S324A,T325A is degraded. The failure of DMiroS182A,S324A,T325A to prevent degradation under this condition might be due to PINK1-mediated phosphorylation on other sites that promote DMiro degradation, or due to activation of additional mechanisms. In two recent studies, MiroS156A is significantly degraded by co-expression of PINK1 and Parkin in addition to CCCP treatment in Hela cells, or by overexpression of Parkin together with Carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP, another mitochondrial uncoupler) treatment in SH-SY5Y cells; whereas in a previous study, MiroS156A is resistant to degradation when only PINK1 or Parkin is individually expressed in HEK293T cells (Wang, 2011). This again suggests that if the PINK1/Parkin pathway is overwhelmingly activated, mutating the few known PINK1-mediated phosphorylation residues in Miro is not sufficient to prevent its degradation (Tsai, 2014).
Why is mitochondrial motility increased in "DMironull, da
>
DMiroS182A,S324A,T325A"? DMiroS182A,S324A,T325A is resistant to PINK1/Parkin-mediated degradation, which may lead to more DMiroS182A,S324A,T325A accumulation on mitochondria. Unexpectedly, DMiroS182A,S324A,T325A protein level in "DMironull, da >
DMiroS182A,S324A,T325A" is not significantly upregulated as compared with DMirowildtype in "DMironull, da
>
DMirowildtype" using fly whole body lysates. It is likely that PINK1/Parkin-dependent degradation of Miro only occurs in certain cell types, at certain subcellular locations, on certain populations of mitochondria, or under certain circumstances, and thus it is hard to detect a dramatic change using whole body lysates or without overexpression of PINK1/Parkin. Future mechanistic study is needed to test these hypotheses, such as detecting Miro subcellular localization and expression levels in different cell types, in different developmental stages, and with different mitochondrial stresses (Tsai, 2014).
This work highlights the importance of a precise control of mitochondrial movement for neuronal health. Anterograde mitochondrial transport in axons is mediated by a conserved motor/adaptor complex, which includes the motor kinesin heavy chain (KHC), the adaptor protein Milton and the mitochondrial membrane anchor Miro. In the current model, Miro binds to Milton, which in turn binds to KHC recruiting mitochondria to the motors and microtubules. In addition to the transmembrane domain inserted into the OMM, Miro features a pair of EF-hands and two GTPase domains. Miro was also recently found to be a substrate of the Ser/Thr kinase PINK1 and of the E3 ubiquitin ligase Parkin, both mutated in PD. Thus, mitochondrial transport can be regulated by multiple signals upstream of Miro and the motor complex maintaining energy and Ca2+ homeostasis in neuronal processes and terminals. For example, loss of PINK1-mediated phosphorylation of DMiro increases local mitochondrial movement at NMJs. In turn, this may disrupt synaptic homeostasis leading to synaptic overgrowth by mechanisms yet to be identified. Similarly, the loss of DA neurons in adult brains could well be a consequence of impaired synaptic homeostasis together with an accumulation of dysfunctional mitochondria. Local signals that regulate mitochondrial transport through Miro must be crucial to supporting neuronal functions. This study elucidates a fundamental biological mechanism demanded by a healthy neuron (Tsai, 2014).
Mitochondrial morphology regulatory proteins interact with signaling pathways involved in differentiation. In Drosophila oogenesis, EGFR signaling regulates mitochondrial fragmentation in posterior follicle cells (PFCs). EGFR driven oocyte patterning and Notch signaling mediated differentiation are abrogated when PFCs are deficient for the mitochondrial fission protein Drp1. It is not known whether fused mitochondrial morphology in drp1 mutant PFCs exerts its effects on these signaling pathways through a change in mitochondrial electron transport chain (ETC) activity. This study shows that aggregated mitochondria in drp1 mutant PFCs have increased mitochondrial membrane potential. Experiments were performed to assess the signaling pathway regulating mitochondrial membrane potential and how this impacts follicle cell differentiation. drp1 mutant PFCs were found to show an increase in phosphorylated ERK (dpERK) formed downstream of EGFR signaling. ERK regulates high mitochondrial membrane potential in drp1 mutant PFCs. PFCs depleted of ERK and drp1 are able to undergo Notch mediated differentiation. Notably mitochondrial membrane potential decrease via ETC inhibition activates Notch signaling at an earlier stage in wild type and suppresses the Notch signaling defect in drp1 mutant PFCs. Thus, this study shows that the EGFR pathway maintains mitochondrial morphology and mitochondrial membrane potential in follicle cells for its functioning and decrease in mitochondrial membrane potential is needed for Notch mediated differentiation (Tomer, 2018).
The Drosophila gene products Bet1, Slh and CG10144, predicted to function in intracellular vesicle trafficking, were previously found to be essential for mitochondrial nucleoid maintenance. This study shows that Slh and Bet1 co-operate to maintain mitochondrial functions. In their absence, mitochondrial content, membrane potential and respiration became abnormal, accompanied by mitochondrial proteotoxic stress, but without direct effects on mtDNA. Immunocytochemistry showed that both Slh and Bet1 are localized at the Golgi, together with a proportion of Rab5-positive vesicles. Some Bet1, as well as a tiny amount of Slh, co-fractionated with highly purified mitochondria, whilst live-cell imaging showed coincidence of fluorescently tagged Bet1 with most Lysotracker-positive and a small proportion of Mitotracker-positive structures. This 3-way association was disrupted in cells knocked down for Slh, although co-localized lysosomal and mitochondrial signals were still seen. Neither Slh nor Bet1 were required for global mitophagy or endocytosis, but prolonged Slh knockdown resulted in G2 growth arrest, with increased cell diameter. These effects were shared with knockdown of betaCOP but not of CG1044, Snap24 or Syntaxin6. These findings implicate vesicle sorting at the cis-Golgi in mitochondrial quality control (Gerards, 2018).
Mitochondrial aging, which results in mitochondrial dysfunction, is strongly linked to many age-related diseases. Aging is associated with mitochondrial enlargement and transport of cytosolic proteins into mitochondria. The underlying homeostatic mechanisms that regulate mitochondrial morphology and function, and their breakdown during aging, remain unclear. This study identified a mitochondrial protein trafficking pathway in Drosophila melanogaster involving the mitochondria-associated protein Dosmit. Dosmit induces mitochondrial enlargement and the formation of double-membraned vesicles containing cytosolic protein within mitochondria. The rate of vesicle formation increases with age. Vesicles originate from the outer mitochondrial membrane as observed by tracking Tom20 localization, and the process is mediated by the mitochondria-associated Rab32 protein. Dosmit expression level is closely linked to the rate of ubiquitinated protein aggregation, which are themselves associated with age-related diseases. The mitochondrial protein trafficking route mediated by Dosmit offers a promising target for future age-related mitochondrial disease therapies (Chen, 2020).
Understanding the molecular mechanisms underlying variation in lifespan is central to ensure long life. Lim3 encoding a homolog of the vertebrate Lhx3/4 transcription factors plays a key role in Drosophila neuron development. This study demonstrated that Lim3 knockdown early in life decreased survival of adult flies. To study the mechanisms underlying this effect, embryonic Lim3 targets were identified using combined RNA-seq and RT-qPCR analyses complemented by in silico analysis of Lim3 binding sites. Though genes with neuronal functions were revealed as Lim3 targets, the characteristics of neurons were not affected by Lim3 depletion. Many of the direct and indirect Lim3 target genes were associated with mitochondrial function, ATP-related activity, redox processes and antioxidant defense. Consistent with the observed changes in the embryonic transcription of these genes, ROS levels were increased in embryos, which could cause changes in the transcription of indirect Lim3 targets known to affect lifespan.It is hypothesized that altered mitochondrial activity is crucial for the decrease of adult lifespan caused by Lim3 knockdown early in life. In adults that encountered Lim3 depletion early in life, the transcription of several genes remained altered, and mitochondrial membrane potential, ATP level and locomotion were increased, confirming the existence of carry-over effects (Rybina, 2019).
An important role of the insect cuticle is to prevent wetting (i.e., permeation of water) and also to prevent penetration of potentially harmful substances. This barrier function mainly depends on the hydrophobic cuticle surface composed of lipids including cuticular hydrocarbons (CHCs). This study investigated to what extent the cuticle inward barrier function depends on the genotype, comprising mitochondrial and nuclear genes in the fruit fly Drosophila melanogaster, and investigated the contribution of interactions between mitochondrial and nuclear genotypes (mito-nuclear interactions) on this function. In addition, the effects of nutrition and sex on the cuticle barrier function were assessed. Based on a dye penetration assay, this study found that cuticle barrier function varies across three fly lines that were captured from geographically separated regions in three continents. Testing different combinations of mito-nuclear genotypes, it was shown that the inward barrier efficiency is modulated by the nuclear and mitochondrial genomes independently. An interaction was found between diet and sex. These findings provide new insights into the regulation of cuticle inward barrier function in nature (Dong, 2019).
Mitochondrial cristae contain electron transport chain (ETC) complexes and are distinct from the inner boundary membrane (IBM). While many details regarding the regulation of mitochondrial structure are known, the relationship between cristae structure and function during organelle development is not fully described. This study used serial-section tomography to characterize the formation of lamellar cristae in immature mitochondria during a period of dramatic mitochondrial development that occurs after Drosophila emergence as an adult. The formation of lamellar cristae was found to be associated with the gain of COX function, and the COX subunit, COX4, was localized predominantly to organized lamellar cristae. Interestingly, 3D tomography showed some COX-positive lamellar cristae were not connected to IBM. It is hypothesized that some lamellar cristae may be organized by a vesicle germination process in the matrix, in addition to invagination of IBM. OXA1 protein, which mediates membrane insertion of COX proteins, was also localized to cristae and reticular structures isolated in the matrix additional to the IBM, suggesting that it may participate in the formation of vesicle germination-derived cristae. Overall, this study elaborates on how cristae morphogenesis and functional maturation are intricately associated. The data support the vesicle germination and membrane invagination models of cristae formation (Jiang, 2019).
Mitochondria are essential organelles that have recently emerged as hubs for several metabolic and signaling pathways in the cell. Mitochondrial morphology is regulated by constant fusion and fission events to maintain a functional mitochondrial network and to remodel the mitochondrial network in response to external stimuli. Although the role of mitochondria in later stages of spermatogenesis has been investigated in depth, the role of mitochondrial dynamics in regulating early germ cell behavior is relatively less-well understood. Previously it was demonstrated that mitochondrial fusion is required for germline stem cell (GSC) maintenance in the Drosophila testis. This study shows that mitochondrial fission is also important for regulating the maintenance of early germ cells in larval testes. Inhibition of Drp1 in early germ cells resulted in the loss of GSCs and spermatogonia due to the accumulation of reactive oxygen species (ROS) and activation of the EGFR pathway in adjacent somatic cyst cells. EGFR activation contributed to premature germ cell differentiation. These data provide insights into how mitochondrial dynamics can impact germ cell maintenance and differentiation via distinct mechanisms throughout development (Senos Demarco, 2019).
Brain stem cells stop dividing in late Drosophila embryos and begin dividing again in early larvae after feeding induces reactivation. Quiescent neural stem cells (qNSCs) display an unusual cytoplasmic protrusion that is no longer present in reactivated NSCs. The protrusions join the qNSCs to the neuropil, brain regions that are thought to maintain NSCs in an undifferentiated state, but the function of the protrusions is not known. This study shows that qNSC protrusions contain clustered mitochondria that are likely maintained in position by slow forward-and-backward microtubule growth. Larvae treated with a microtubule-stabilizing drug show bundled microtubules and enhanced mitochondrial clustering in NSCs, together with reduced qNSC reactivation. It was further shown that intestinal stem cells contain mitochondria-enriched protrusions. The qNSC and intestinal stem-cell protrusions differ from previously reported cytoplasmic extensions by forming stem-cell-to-niche mitochondrial bridges that could potentially both silence genes and sense signals from the stem cell niche (Endow, 2019).
Gut microbiota have been shown to promote oogenesis and fecundity, but the mechanistic basis of remote influence on oogenesis remained unknown. This study reports a systemic mechanism of influence mediated by bacterial-derived supply of mitochondrial coenzymes. Removal of microbiota decreased mitochondrial activity and ATP levels in the whole-body and ovary, resulting in repressed oogenesis. Similar repression was caused by RNA-based knockdown of mitochondrial function in ovarian follicle cells. Reduced mitochondrial function in germ-free (GF) females was reversed by bacterial recolonization or supplementation of riboflavin, a precursor of FAD and FMN. Metabolomics analysis of GF females revealed a decrease in oxidative phosphorylation and FAD levels and an increase in metabolites that are degraded by FAD-dependent enzymes (e.g., amino and fatty acids). Riboflavin supplementation opposed this effect, elevating mitochondrial function, ATP, and oogenesis. These findings uncover a bacterial-mitochondrial axis of influence, linking gut bacteria with systemic regulation of host energy and reproduction (Gnainsky, 2021).
Intracellular temperature affects a wide range of cellular functions in living organisms. However, it remains unclear whether temperature in individual animal cells is controlled autonomously as a response to fluctuations in environmental temperature. Using two distinct intracellular thermometers, this study found that the intracellular temperature of steady-state Drosophila S2 cells is maintained in a manner dependent on Δ9-fatty acid desaturase DESAT1, which introduces a double bond at the Δ9 position of the acyl moiety of acyl-CoA. The DESAT1-mediated increase of intracellular temperature is caused by the enhancement of F(1)F(o)-ATPase-dependent mitochondrial respiration, which is coupled with thermogenesis. This study also revealed that F(1)F(o)-ATPase-dependent mitochondrial respiration is potentiated by cold exposure through the remodeling of mitochondrial cristae structures via DESAT1-dependent unsaturation of mitochondrial phospholipid acyl chains. Based on these findings, a cell-autonomous mechanism is proposed for intracellular temperature control during environmental temperature changes (Murakami, 2022).
Mitochondrial ribosomal proteins (MRPs) assemble as specialized ribosome to synthesize mtDNA-encoded proteins, which are essential for mitochondrial bioenergetic and metabolic processes. MRPs are required for fundamental cellular activities during animal development, but their roles beyond mitochondrial protein translation are poorly understood. This study reports a conserved role of the mitochondrial ribosomal protein L4 (mRpL4) in Notch signaling. Genetic analyses demonstrate that mRpL4 is required in the Notch signal-receiving cells to permit target gene transcription during Drosophila wing development. mRpL4 physically and genetically interacts with the WD40 repeat protein Wings apart (Wap) and activates the transcription of Notch signaling targets. This study shows that human mRpL4 is capable of replacing fly mRpL4 during wing development. Furthermore, knockout of mRpL4 in zebrafish leads to downregulated expression of Notch signaling components. Thus, this study has discovered a previously unknown function of mRpL4 during animal development (Mo, 2023).
Miro GTPases control mitochondrial morphology, calcium homeostasis, and regulate mitochondrial distribution by mediating their attachment to the kinesin and dynein motor complex. It is not clear, however, how Miro proteins spatially and temporally integrate their function as acute disruption of protein function has not been performed. To address this issue, an optogenetic loss of function "Split-Miro" allele was developed for precise control of Miro-dependent mitochondrial functions in Drosophila. Rapid optogenetic cleavage of Split-Miro leads to a striking rearrangement of the mitochondrial network, which is mediated by mitochondrial interaction with the microtubules. Unexpectedly, this treatment did not impact the ability of mitochondria to buffer calcium or their association with the endoplasmic reticulum. While Split-Miro overexpression is sufficient to augment mitochondrial motility, sustained photocleavage shows that Split-Miro is surprisingly dispensable to maintain elevated mitochondrial processivity. In adult fly neurons in vivo, Split-Miro photocleavage affects both mitochondrial trafficking and neuronal activity. Furthermore, functional replacement of endogenous Miro with Split-Miro identifies its essential role in the regulation of locomotor activity in adult flies, demonstrating the feasibility of tuning animal behaviour by real-time loss of protein function (Mattedi, 2023).
Intrapopulation variation in behaviour, including activity, boldness and aggressiveness, is becoming more widely recognized and is hypothesized to substantially affect ecological and evolutionary dynamics. Although previous studies used candidate-gene approaches and genome-wide association analyses to identify genes correlated with variations in activity and aggressiveness, behavioural variation may not be fully captured in the nuclear genome, as it does not account for mitochondrial genomes. Mitochondrial genes encode products that are key regulators of the cellular energy-producing pathways in metabolic processes and are thought to play a significant role in life-history and reproductive traits. This study considered many isofemale lines of Drosophila immigrans established from two wild populations to investigate whether intrapopulation variation in the mitochondrial genome affected activity level within this species. Two major haplogroups in these populations, and activity levels in both larvae and adults differed significantly between the two haplogroups. This result indicated that intrapopulation variation in activity level may be partially controlled by mitochondrial genes, along with the interaction between nuclear and mitochondrial genes and the age of individual organisms (Ueno, 2021).
While mitochondria have long been understood to be critical to cellular function, questions remain as to how genetic variation within mitochondria may underlie variation in general metrics of organismal function. To date, studies investigating links between mitochondrial genotype and phenotype have largely focused on differences in expression of genes and physiological and life-history traits across haplotypes. Mating display behaviours may also be sensitive to mitochondrial functionality and so may also be affected by sequence variation in mitochondrial DNA, with consequences for sexual selection and fitness. This study tested whether the pre-copulatory mating success of male fruit flies (Drosophila melanogaster) varies across six different mitochondrial haplotypes expressed alongside a common nuclear genetic background. A significant effect of mitochondrial haplotype was found on measuring of competitive mating success, driven largely by the relatively poor performance of males with one particular haplotype. This haplotype, termed 'Brownsville', has previously been shown to have complex and sex-specific effects, most notably including depressed fertility in males but not females. This study extends this disproportionate effect on male reproductive success to pre-copulatory aspects of reproduction. The results demonstrate that mutations in mitochondrial DNA can plausibly affect pre-copulatory mating success, with implications for future study into the subcellular underpinnings of such behaviours and the information they may communicate (Koch, 2022).
Reactive oxygen species (ROS) and mitochondrial defects in neurons are implicated in neurodegenerative disease. This study finds that a
key consequence of ROS and neuronal mitochondrial dysfunction is the
accumulation of lipid droplets (LD) in glia. In Drosophila, ROS triggers c-Jun-N-terminal Kinase (JNK) and Sterol Regulatory
Element Binding Protein (SREBP) activity in neurons leading to
LD accumulation in glia prior to or at the onset of neurodegeneration. The accumulated lipids were peroxidated in the presence of ROS. Reducing LD accumulation in glia and lipid peroxidation via targeted lipase overexpression and/or
lowering ROS significantly delayed the onset of neurodegeneration.
Furthermore, a similar pathway led to glial LD accumulation in Ndufs4
mutant mice with neuronal mitochondrial defects, suggesting that LD
accumulation following mitochondrial dysfunction is an
evolutionarily conserved phenomenon, and represents an early,
transient indicator and promoter of neurodegenerative disease (Liu, 2015).
This study shows that neuronal mitochondrial defects that lead to elevated levels of ROS, induce activation of JNK and SREBP, which in turn elevate lipid synthesis in neurons and formation of LD in glial cells. These LDs contribute to and promote ND through elevated levels of lipid peroxidation. LDs form in glia prior to or at the onset of the appearance of obvious degenerative histological features in Drosophila and mice. Reducing the number and size of LD pharmacologically or genetically delays ND in the fly. This is the first indication that SREBP, lipid droplet biogenesis, and lipid metabolism play a role in the pathogenesis of several neurodegenerative diseases (Liu, 2015).
A growing body of evidence points to the importance of glial health and function in nervous system energy metabolism and homeostasis. Nevertheless, given the number and prevalence of different types of neurodegenerative diseases, very few reports have documented the presence of LDs in either neuron or glia in patients and in animal models. LD accumulation in the brain has been reported in cells that line the ventricles in the globus pallidus and substantia nigra in mutant mice lacking both subunits of the liver X receptor, apolipoprotein E, or a peroxisomal biogenesis factor (Pex5) . In addition, in vitro studies using immortalized cell lines and explants show that LD may form and accumulate in glia under conditions of nutrient deprivation or lipopolysaccharide induced stress. However, LDs have not been shown to play an active role in neurodegenerative processes. Furthermore, LD accumulation has not been reported in patients with or animal models of Leigh syndrome (NDUFS4/Ndufs4, NDUFAF6/sicily), CMT-2A2 or HMSN6 (MFN2/Marf), and ARSAL (MARS2/Aats-met). The lack of neuropathological reports of LDs in animal models or in patients with ND may be attributed to the fact that LD accumulation is transient and mostly occur during presymptomatic stages of the disease (Liu, 2015).
Although these genes/mutants are implicated in very different mitochondrial processes, they exhibit a common phenotype of elevated levels of ROS, leading to LD accumulation. Similar morphological changes of glia have been reported under stress conditions. Interestingly, mid- and late-stage Ndufs4-/- mice exhibit CNS lesions in the same brain regions where the LD accumulate in early stage animals, showing a strong correlative relationship. Similarly, LD accumulation in Drosophila mutants occurs prior to or at the onset of physical signs of ND. Importantly, the delivery of AD4 is able to significantly ameliorate LD accumulation in Drosophila and delay the onset of ND in flies and mice. Hence, the molecular mechanisms underlying these phenotypes are likely to be conserved between these species and potentially also in higher organisms (Liu, 2015).
In the clinical setting, the prescription of antioxidants toward treatment of neurodegenerative diseases has been tested repeatedly on patients with neurodegenerative disorders, without compelling results. The LD accumulation phenotype in these mutants occurs prior to histopathological and physical signs of ND. A brief period of AD4 delivery prior to the onset of symptoms in mutant mice is effective in delaying onset of clinical signs. Thus, therapy with an effective antioxidant that penetrates the blood-brain barrier should be started early and sustained over long periods. In addition, pharmacological manipulation of JNK or lipid levels in the brain may serve as a potential therapy to delay the onset of ND. However, similar to antioxidant treatment, this may need to be administered at an early stage. Hence, early identification of potential ROS related neurological disease based on genetic/genomic diagnosis or by biomarkers may be critical. Since LD accumulation is one of the earliest presymptomatic changes that occurs in the nervous system, detection of LD itself or changes in neurometabolism may be a promising biomarker (Liu, 2015).
In summary, this study provides evidence for the role of altered lipid metabolism and a neuron-glia interplay that promotes ND. In some mitochondrial mutants, an upregulation of SREBP was observed, as well as lipid biogenesis and glial LD formation. The accumulation of LD is not sufficient to promote the ND process itself. However, in the presence of ROS the accumulated lipids are peroxidated and promote ND, possibly by promoting the release of lipids from LD, elevating the cytoplasmic load, and causing a progressive loss of LD. Hence, the synergistic effects of increased lipid synthesis and/or LD accumulation in combination with elevated ROS and lipid peroxidation promote ND. Finally, it was shown that LD accumulation occurs at the onset or precedes ND in flies and mice, suggesting that LD and changes in lipid metabolism in the nervous system may be a promising biomarker to identify brain regions susceptible to but not yet exhibiting symptoms of ND (Liu, 2015).
Previous studies have established the anticancer effect of vitamin K2 (VK2). However, its effect on lymphoma induced by UBIAD1/heix mutation in Drosophila remains unknown. Therefore, this study aimed to develop an in vivo model of lymphoma for the precise characterization of lymphoma phenotypes. This study also aimed to improve the understanding of the mechanisms that underlie the preventative effects of VK2 on lymphoma. The results demonstrated that VK2 prevents lymphoma by acting as an electron carrier and by correcting the function and structure of mitochondria by inhibiting mitochondrial reactive oxygen species production mtROS. This work identifies mitochondria as a key player in cancer therapy strategies (Dragh, 2017).
Most differentiated cells convert glucose to pyruvate in the cytosol through glycolysis, followed by pyruvate oxidation in the mitochondria. These processes are linked by the mitochondrial pyruvate carrier (MPC), which is required for efficient mitochondrial pyruvate uptake. In contrast, proliferative cells, including many cancer and stem cells, perform glycolysis robustly but limit fractional mitochondrial pyruvate oxidation. This study sought to understand the role this transition from glycolysis to pyruvate oxidation plays in stem cell maintenance and differentiation. Loss of the MPC in Lgr5-EGFP-positive stem cells, or treatment of intestinal organoids with an MPC inhibitor, increases proliferation and expands the stem cell compartment. Similarly, genetic deletion of the MPC in Drosophila intestinal stem cells also increases proliferation, whereas MPC overexpression suppresses stem cell proliferation. These data demonstrate that limiting mitochondrial pyruvate metabolism is necessary and sufficient to maintain the proliferation of intestinal stem cells (Schell, 2017).
It was first observed almost 100 years ago that, unlike differentiated cells, cancer cells tend to avidly consume glucose, but not fully oxidize the pyruvate that is generated from glycolysis. This was originally proposed to be due to dysfunctional or absent mitochondria, but it has become increasingly clear that mitochondria remain functional and critical. Mitochondria are particularly important in proliferating cells because essential steps in the biosynthesis of amino acids, nucleotide and lipid occur therein. Most proliferating stem cell populations also exhibit a similar glycolytic metabolic program, which transitions to a program of mitochondrial carbohydrate oxidation during differentiation. The first distinct step in carbohydrate oxidation is import of pyruvate into the mitochondrial matrix, where it gains access to the pyruvate dehydrogenase complex (PDH) and enters the tricarboxylic acid (TCA) cycle as acetyl-CoA. The two proteins that assemble to form the mitochondrial pyruvate carrier (MPC) have been recently described. This complex is necessary and sufficient for mitochondrial pyruvate import in yeast, flies and mammals, and thereby serves as the junction between cytoplasmic glycolysis and mitochondrial oxidative phosphorylation. Decreased expression and activity of the MPC underlies the glycolytic program in colon cancer cells in vitro, and forced re-expression of the MPC subunits increased carbohydrate oxidation and impaired the ability of these cells to form colonies in vitro and tumours in vivo. This impairment of tumorigenicity was coincident with a loss of key markers and phenotypes associated with stem cells. This has prompted an examination of whether glycolytic non-transformed stem cells might also exhibit low MPC expression and mitochondrial pyruvate oxidation, which must increase during differentiation (Schell, 2017).
The role of the MPC was studied in the ISCs of the fruit fly Drosophila, which share key aspects of their biology with mammalian ISCs. Both MPC1 and MPC2 are expressed in all four cell types of the intestine, with the lowest level of expression in the ISCs and the highest expression in the differentiated enteroendocrine cells. Confocal imaging of intestines dissected from dMPC1 mutants revealed that the epithelium exhibits multilayering unlike the normal single-cell layer seen in controls. This is a classic overgrowth phenotype that is associated with oncogene mutations in Drosophila. Accordingly, MARCM clonal analysis was used to determine whether a specific loss of the MPC in ISCs leads to an increase in their proliferation. On average, newly divided GFP-marked dMPC1 mutant clones are more than twofold larger than control clones, which were generated in parallel using a wild-type chromosome, indicating that the MPC is required in the ISC lineage to suppress proliferation. Because GFP-marked clones could include cells that differentiate into mature enterocytes or enteroendocrine cells, clonal analysis was conducted in the absence of Notch, thereby blocking ISC differentiation. Under these conditions, an approximately twofold increase was observed in the size of dMPC1 mutant ISC clones. To confirm and extend these results, MPC function was specifically disrupted in the ISCs by using the Dl-GAL4 driver in combination with UAS-GFP, which facilitates stem cell identification. Once again, approximately twofold more GFP-marked stem cells were observed relative to controls when either dMPC1 or dMPC2 expression was disrupted by RNA-mediated interference (RNAi) along with increased ISC proliferation as detected by staining for phosphorylated histone H3 (pHH3). Similar results were obtained when RNAi was targeted to the E1 or E2 subunits of PDH to specifically disrupt the next step in mitochondrial pyruvate oxidation. Importantly, an opposite phenotype was seen when Ldh was reduced by RNAi in the ISCs or progenitor cells. Ldh suppression is known to result in a significant increase in pyruvate levels, which can promote pyruvate oxidation. Taken together with the results with Pdh RNAi, these observations support the model that the MPC limits stem cell proliferation through promoting oxidative pyruvate metabolism in the mitochondria. It also appears to be sufficient as specific overexpression of MPC1 and MPC2 in ISCs or progenitors caused a reduction in stem cell proliferation, the opposite of the loss-of-function phenotype. This can be seen in either Pseudomonas-infected intestines, which undergo rapid stem cell proliferation, or under basal conditions in aged animals. Consistent with this, MPC overexpression under basal conditions had no effects on intestinal morphology, while the intestines from infected flies displayed a fully penetrant size reduction, which is probably due to the inability of ISCs to maintain tissue homeostasis. Taken together, these results demonstrate that mitochondrial pyruvate uptake and metabolism is both necessary and sufficient in a stem cell autonomous manner to regulate ISC proliferation and maintain intestinal homeostasis in Drosophila (Schell, 2017).
Studies in Drosophila, intestinal organoids and mice provide strong evidence that the MPC is necessary and sufficient in a cell autonomous manner to suppress stem cell proliferation. Consistently, this study has demonstrated that ISCs maintain low expression of the subunits that comprise the MPC, which enforces a mode of carbohydrate metabolism wherein glucose is metabolized in the cytosol to pyruvate and other biosynthetic intermediates. This glycolytic metabolic program appears to be sufficient to drive robust and continuous stem cell proliferation. High mitochondrial content was observed in ISCs, which must be geared primarily toward biosynthetic functions and/or oxidation of other substrates such as fatty acids. Increased fatty acids, the metabolism of which is enhanced in MPC-deficient and MPC-inhibited organoids, have been shown to promote ISC expansion and proliferation via enhanced beta-catenin signalling and increasing tumour-initiating capacity. MPC expression increases following differentiation, consistent with the shift in demand from macromolecule biosynthesis to ATP production in support of post-mitotic differentiated cell function. A similar switch in MPC expression can be seen following differentiation of embryonic stem cells, haematopoietic stem cells and trophoblast stem cells. Conversely, MPC expression is reduced after reprogramming fibroblasts to induced pluripotent stem cells. This suggests that the effects of altering pyruvate flux that wad observed in this study might not be restricted to ISCs, but instead be representative of similar effects on multiple stem cell populations. Interestingly, Myc is known to drive a metabolic program that is similar to that observed following MPC loss, characterized by increased glycolysis and reliance on glutamine and fatty acid oxidation with reduced glucose oxidation. This suggests that Myc may play a role in repressing the MPC in stem cells, possibly acting downstream of Wnt/beta-catenin signalling. Consistent with this, Myc and its repressive co-factors localize to the Mpc1 promoter and Myc expression is strongly anti-correlated with Mpc1 expression (Schell, 2017).
Taken together, these studies demonstrate that changes in the MPC and mitochondrial pyruvate metabolism are required to properly orchestrate the proliferation and homeostasis of intestinal stem cells. Importantly, this metabolic program, mediated at least partially by the MPC, appears to be instructive for cell fate, rather than a downstream consequence of cell fate. Future work will define the extent to which the results presented in this study relate to those showing that diet quality and quantity can modulate ISC behaviour. It is tempting to speculate that ISC metabolism is used as a signal for increased or decreased demand for intestinal epithelium. Perhaps of most importance will be to define the mechanisms whereby altered partitioning of pyruvate metabolism affects stem cell proliferation and fate. It is speculated that the robust changes that were observed in fatty acid oxidation and histone acetylation, which are probably downstream of altered metabolite utilization for acetyl-CoA production, play an important role. While the mechanisms are not as yet defined, these studies establish a paradigm wherein mitochondrial metabolism does not merely provide a permissive context for proliferation or differentiation, but rather plays a direct and instructive role in controlling stem cell fate (Schell, 2017).
Iron sulfur (Fe-S) clusters and the molybdenum cofactor (Moco) are present at enzyme sites, where the active metal facilitates electron transfer. Such enzyme systems are soluble in the mitochondrial matrix, cytosol and nucleus, or embedded in the inner mitochondrial membrane, but virtually absent from the cell secretory pathway. They are of ancient evolutionary origin supporting respiration, DNA replication, transcription, translation, the biosynthesis of steroids, heme, catabolism of purines, hydroxylation of xenobiotics, and cellular sulfur metabolism. RNA interference of Mocs3 disrupts Moco biosynthesis and the circadian clock. Fe-S-dependent mitochondrial respiration is discussed in the context of germ line and somatic development, stem cell differentiation and aging. The subcellular compartmentalization of the Fe-S and Moco assembly machinery components and their connections to iron sensing mechanisms and intermediary metabolism are emphasized. A biochemically active Fe-S core complex of heterologously expressed fly Nfs1, Isd11, IscU, and human frataxin is presented. Based on the recent demonstration that copper displaces the Fe-S cluster of yeast and human ferredoxin, an explanation for why high dietary copper leads to cytoplasmic iron deficiency in flies is proposed. Another proposal that exosomes contribute to the transport of xanthine dehydrogenase from peripheral tissues to the eye pigment cells is put forward, where the Vps16a subunit of the HOPS complex may have a specialized role in concentrating this enzyme within pigment granules. Finally, a hypothesis is formulated that (1) mitochondrial superoxide mobilizes iron from the Fe-S clusters in aconitase and succinate dehydrogenase; (2) increased iron transiently displaces manganese on superoxide dismutase, which may function as a mitochondrial iron sensor since it is inactivated by iron; (3) with the Krebs cycle thus disrupted, citrate is exported to the cytosol for fatty acid synthesis, while succinyl-CoA and the iron are used for heme biosynthesis; (4) as iron is used for heme biosynthesis its concentration in the matrix drops allowing for manganese to reactivate superoxide dismutase and Fe-S cluster biosynthesis to reestablish the Krebs cycle (Marelja, 2018).
Seipin, the gene that causes Berardinelli-Seip congenital lipodystrophy type 2 (BSCL2), is important for adipocyte differentiation and lipid homeostasis. Previous studies in Drosophila revealed that Seipin promotes ER calcium homeostasis through the Ca(2+)-ATPase SERCA, but little is known about the events downstream of perturbed ER calcium homeostasis that lead to decreased lipid storage in Drosophila dSeipin mutants. This study shows that glycolytic metabolites accumulate and the downstream mitochondrial TCA cycle is impaired in dSeipin mutants. The impaired TCA cycle further leads to a decreased level of citrate, a critical component of lipogenesis. Mechanistically, Seipin/SERCA-mediated ER calcium homeostasis is important for maintaining mitochondrial calcium homeostasis. Reduced mitochondrial calcium in dSeipin mutants affects the TCA cycle and mitochondrial function. The lipid storage defects in dSeipin mutant fat cells can be rescued by replenishing mitochondrial calcium or by restoring the level of citrate through genetic manipulations or supplementation with exogenous metabolites. Together, these results reveal that Seipin promotes adipose tissue lipid storage via calcium-dependent mitochondrial metabolism (Ding, 2018).
Impaired lipid metabolism is associated with an imbalance in energy homeostasis and many other disorders. Excessive lipid storage results in obesity, while a lack of adipose tissue leads to lipodystrophy. Clinical investigations reveal that obesity and lipodystrophy share some common secondary effects, especially non-alcoholic fatty liver disease and severe insulin resistance. Berardinelli-Seip congenital lipodystrophy type 2 (BSCL2/CGL2) is one of the most severe lipodystrophy diseases. Patients with BSCL2 manifest almost total loss of adipose tissue as well as fatty liver, insulin resistance, and myohypertrophy. BSCL2 results from mutation of the Seipin gene, which is highly conserved from yeast to human (Ding, 2018).
To study the function of Seipin, genetic models were established in different organisms, including yeast, fly, and mouse, and in human cells. As a transmembrane protein residing in the endoplasmic reticulum (ER) and in the vicinity of lipid droplet (LD) budding sites, Seipin has been shown to be involved in LD formation, phospholipid metabolism, lipolysis, and ER calcium homeostasis. As a result of the functional studies in these models, several factors that interact with Seipin protein were identified, such as the phosphatidic acid phosphatase lipin, 14-3-3β, and glycerol-3-phosphate acyltransferase (GPAT). Drosophila Seipin (dSeipin) functions tissue autonomously in preventing ectopic lipid accumulation in salivary gland (a non-adipose tissue) and in promoting lipid storage in fat tissue (Tian, 2011). The non-adipose tissue phenotype is likely attributed to the increased level of phosphatidic acid (PA) generated by elevated GPAT activity (Pagac, 2016). In adipose tissue Seipin interacts with the ER Ca2+-ATPase SERCA, whose activity is reduced in dSeipin mutants, leading to reduced ER calcium levels. Further genetic analysis suggested that the perturbed level of intracellular calcium contributes to the lipodystrophy. However, it is not known how the depleted ER calcium pool causes decreased lipid storage (Ding, 2018).
Besides the ER, mitochondria are another important intracellular calcium reservoir. Mitochondrial calcium is mainly derived from the ER through the IP3R channel. IP3R not only releases calcium from the ER into the cytosol, but also provides sufficient Ca2+ at mitochondrion-associated ER membranes (MAMs) for activation of the mitochondrial calcium uniporter. The mitochondrial Ca2+ level varies greatly in different cell types and can be modulated by influx and efflux channel proteins, such as MCU and NCLX, a mitochondrial Na+/Ca2+ exchanger. A proper mitochondrial Ca2+ level is implicated in mitochondrial integrity and function. Mitochondrial calcium is needed to support the activity of the mitochondrial matrix dehydrogenases in the TCA cycle. TCA cycle intermediates are used for the synthesis of important compounds, including glucose, amino acids, and fatty acids. Acetyl-CoA, as the basic building block of fatty acids, is generally derived from glycolysis, the TCA cycle, and fatty acid β-oxidation. In mammalian adipocytes, acetyl-CoA derived from the TCA cycle intermediate citrate is crucial for de novo lipid biosynthesis, which contributes significantly to lipid storage (Ding, 2018 and references therein).
This study used multiple comparative omics to analyze the proteomic, transcriptomic, and metabolic differences between larval fat cells of dSeipin mutants and wild type. The results reveal an impairment in channeling glycolytic metabolites to mitochondrial metabolism in dSeipin mutant fat cells, and scarcity of mitochondrial Ca2+, are the causative factors of this metabolic dysregulation. Evidence is provided showing that dSeipin lipodystrophy is rescued by restoring mitochondrial calcium or replenishing citrate. It is proposed that the low ER Ca2+ level in dSeipin mutants cannot maintain a sufficiently high mitochondrial Ca2+ concentration to support the TCA reactions. This in turn leads to reduced lipogenesis in dSeipin mutants (Ding, 2018).
Seipin promotes fat tissue lipid storage via calcium-dependent mitochondrial metabolism. Defective ER calcium homeostasis in dSeipin mutants is associated with reduced mitochondrial calcium and impaired mitochondrial function, such as low production of TCA cycle metabolites. Restoring mitochondrial calcium levels or replenishing citrate, a key TCA cycle product and also an important precursor of lipogenesis, rescues the lipid storage defects in dSeipin mutant fat cells (Ding, 2018).
This study investigated the underlying causes of Seipin-dependent lipodystrophy by integrating multiple omic analyses, including RNA-seq, quantitative proteomics, and metabolomic analysis. Compared to previous studies based on genetics and traditional cellular phenotypic analysis, these combinatory omic approaches provide an unprecedented spectrum of molecular phenotypes, which not only add new information but also pinpoint logical directions for further investigations (Ding, 2018).
Omics analyses, in particular lipidomic analysis, have been utilized to investigate the underlying mechanisms in several previous Seipin studies and led to the finding that PA is elevated in several Seipin mutant models. In this study, based on genetic rescuing assays and quantitative proteomics analysis, it was initially proposed that downregulated glycolysis is the cause of lipodystrophy. However, both the RNA-seq results and metabolomic data argue against this possibility and suggest a new mechanism. Despite reduced levels of glycolytic enzymes, transcription of the corresponding genes is not affected, and glycolytic metabolites, in particular pyruvate, are increased in dSeipin mutants compared to wild type. Metabolomic data further show that citrate and isocitrate, which are the products of the first two steps of the mitochondrial TCA cycle, are dramatically decreased in dSeipin mutants, suggesting a defective metabolic flow downstream of pyruvate. These results lead to a new possibility that the lipid storage defects in dSeipin mutants are caused by a defective TCA cycle and this is indeed supported by the metabolic flux analysis. These findings further suggest the involvement of mitochondria. In line with this, the previous discovery that fatty acid β-oxidation is elevated in dSeipin mutant fat cells may reflect compensation for the reduced TCA cycle and lipogenesis. This possibility is supported by the results of genetic and citrate-supplement rescue experiments and by citrate measurements (Ding, 2018).
It is known that glycolytic enzymes and metabolites are regulated by a metabolic feedback loop, which may complicate the explanation of genetic interactions. The current findings highlight that although genetic analysis and rescue results provide important clues, multiple lines of evidence are critical for unraveling complex intracellular pathways. In this case, the combination of omic results and genetic analysis led to the finding that mitochondrial metabolism is important in Seipin-associated lipodystrophy (Ding, 2018).
Mitochondria are hubs in key cellular metabolic processes, including the TCA cycle, ATP production, and amino acid catabolism. Mitochondria also play a central role in lipid homeostasis by controlling two seemingly opposite metabolic pathways, lipid biosynthesis, and fatty acid breakdown. Therefore, impairment of mitochondrial function in different tissues may lead to different, even opposite, phenotypes in lipid storage. In tissues where lipid biosynthesis is the major pathway, defective mitochondria might result in reduced lipid storage, whereas in tissues where fatty acid oxidation prevails, the same defect might lead to increased lipid storage. Reduced lipid storage in dSeipin mutants suggests the former case. The reduced level of citrate and other TCA cycle products in dSeipin mutants suggests an impairment of mitochondrial function. The reduction of OCR and ATP production, the decreased Rhod-2 staining, and the aberrant enrichment of mitochondria within autophagosomes all further support this notion. Interestingly, in mouse brown adipose tissue, Seipin mutation increases mitochondrial respiration along with normal MitoTracker labeling (Zhou, 2016). The discrepancies suggest that Seipin may have cell type-specific functions. Unlike white adipose tissue, which favors lipid storage/biosynthesis, brown adipose tissue is prone to fatty acid breakdown (Ding, 2018).
The link between mitochondria and Seipin was concealed in several previous studies. GPATs, which are recently reported Seipin-interacting proteins, participate in many mitochondrial processes. For example, mitochondria from brown adipocytes that are deficient in GPAT4 exhibit high oxidative levels, and mitochondrial GPAT is required for mitochondrial dynamics. PA, which is elevated in Seipin mutants, is required for mitochondrial morphology and function. Similarly, mitochondrial impairments were also observed in various lipodystrophic conditions. Downregulation of mitochondrial transcription and altered mitochondrial function were indicated in type III congenital generalized lipodystrophy. Multiple mitochondrial metabolic processes are altered in mice with lipodystrophy caused by Zmpste24 mutation. HIV patients treated with anti-retroviral therapy manifest partial lipodystrophy and impaired mitochondria in adipocytes. Moreover, mitochondrial dysfunction in adipose tissue triggers lipodystrophy and systemic disorders in mice. Therefore, the contribution of mitochondrial dysfunction to the cause or development of lipodystrophic conditions warrants further examination (Ding, 2018).
It has been previously reported that dSeipin/SERCA-mediated ER calcium homeostasis is critical for lipid storage (Bi, 2014). Consistent with this, transcripts encoding calcium signaling factors are enriched in the genes that are differentially expressed between dSeipin mutants and wild type. Mitochondrial calcium is transported from the ER through the ER-resident channel IP3R. The reduction of mitochondrial calcium in dSeipin mutant fat cells suggests that the decreased ER calcium leads to an insufficient level of mitochondrial calcium. Importantly, RNAi of a putative Drosophila mitochondrial calcium efflux channel (NCLX/CG18660) not only restores the mitochondrial calcium level but also rescues the lipid storage defects in dSeipin mutants, indicating that mitochondrial calcium is key for dSeipin-mediated lipid storage. This explains the previous finding that the lipid storage defects in dSeipin mutants are rescued by RNAi of RyR, which is not required for ER-mitochondrion calcium transport, but not by RNAi of IP3R (Ding, 2018).
Cellular calcium has been linked to lipid storage and related diseases in recent studies. Comprehensive genetic screening in Drosophila showed that ER calcium-related proteins are key regulators of lipid storage. In particular, SERCA, as the sole ER calcium influx channel and an interacting partner of Seipin, has been repeatedly implicated in lipid metabolism. Dysfunctional lipid metabolism can disrupt ER calcium homeostasis by inhibiting SERCA and further disturbing systemic glucose homeostasis. Increased SERCA expression was shown to have dramatic anti-diabetic benefits in mouse models. In a genomewide association study, SERCA was been found to be associated with obesity. In addition, cellular calcium influx is important for transcriptional programming of lipid metabolism, including lipolysis in mice. The current study further elucidates that ER calcium and mitochondrial calcium are important for cellular lipid homeostasis. It also provides a new insight into the pathogenic mechanism of congenital lipodystrophy (Ding, 2018).
Since Seipin mutations lead to opposite effects on lipid storage in adipose tissue (lipodystrophy) and non-adipose tissues (ectopic lipid storage), numerous studies have been carried out to understand the underlying mechanisms. In Seipin mutants, elevated GPAT activity leads to an increased level of PA. This may cause the formation of supersized lipid droplets in non-adipose cells because of the fusogenic property of PA in lipid leaflets, and may also lead to adipogenesis defects due to the potential role of PA as an inhibitor of preadipocyte differentiation. The Seipin-mediated lipid storage phenotype is further complicated by the role of Seipin in lipid droplet formation, which is mainly studied in unicellular eukaryotic yeast or in cultured cells from multicellular eukaryotic organisms. Seipin has been found in the ER-LD contact sites, which are considered as essential subcellular foci for LD formation/maturation. Moreover, in mammalian adipose tissue, the role of Seipin in lipogenesis or lipolysis may also be masked by the defect in early adipogenesis (Ding, 2018).
How can previous findings in different model organisms and different cell types be reconciled? Seipin has been characterized as a tissue-autonomous lipid modulator. It is likely that Seipin participates in lipid metabolism via distinct mechanisms in different tissues. Alternatively, the metabolic processes that involve Seipin may have different outcomes in different tissues. For example, mitochondria have a different impact on lipid metabolism in different tissues: In non-fat cells, mitochondria mainly direct energy mobilization, whereas in fat cells, mitochondria mainly lead anabolism. The molecular role of Seipin and the phenotypic outcomes in Seipin mutants may rely on specific cellular and developmental contexts (Ding, 2018).
This study found that the fundamental scaling relationship between mass and metabolic rate, as well as the oxidative capacity per mitochondria, were found to change significantly across development in the fruit fly Drosophila. However, mitochondrial respiration rate was maintained at similar levels across development. Furthermore, larvae clustered into two types-those that switched to aerobic, mitochondrial ATP production before the second instar and those that relied on anaerobic, glycolytic production of ATP through the second instar. Despite genetic variation for the timing of this metabolic shift, metabolic rate in second-instar larvae was more robust to genetic variation than was the metabolic rate of other instars. Larvae were found with a mitochondrial-nuclear incompatibility that disrupts mitochondrial function had increased aerobic capacity and relied more on anaerobic ATP production throughout development relative to larvae from wild-type strains. By taking advantage of both ways of making ATP, larvae with this mitochondrial-nuclear incompatibility maintained mitochondrial respiratory capacity, but also had higher levels of whole-body reactive oxygen species and decreased mitochondrial membrane potential, potentially as a physiological defense mechanism. Thus, genetic defects in core physiology can be buffered at the organismal level via physiological plasticity and natural populations may harbor genetic variation for distinct metabolic strategies in development that generate similar organismal outcomes (Matoo, 2018).
Mitochondrial Ca(2+) uptake is an important mediator of metabolism and cell death. Identification of components of the highly conserved mitochondrial Ca(2+) uniporter has opened it up to genetic analysis in model organisms. This study reports a comprehensive genetic characterization of all known uniporter components conserved in Drosophila. While loss of pore-forming MCU or EMRE abolishes fast mitochondrial Ca(2+) uptake, this results in only mild phenotypes when young, despite shortened lifespans. In contrast, loss of the MICU1 gatekeeper is developmentally lethal, consistent with unregulated Ca(2+) uptake. Mutants for the neuronally restricted regulator MICU3 are viable with mild neurological impairment. Genetic interaction analyses reveal that MICU1 and MICU3 are not functionally interchangeable. More surprisingly, loss of MCU or EMRE does not suppress MICU1 mutant lethality, suggesting that this results from uniporter-independent functions. The data reveal the interplay among components of the mitochondrial Ca(2+) uniporter and shed light on their physiological requirements in vivo (Tufi, 2019).
The uptake of Ca2+ into mitochondria has long been established as a key regulator of an array of cellular homeostatic processes as diverse as bioenergetics and cell death. A series of seminal discoveries has elucidated the identity of the components that make up the mitochondrial Ca2+ uniporter complex. The mammalian uniporter is composed of MCU (mitochondrial calcium uniporter) as the main pore-forming protein; its paralog MCUb; a small structural component, EMRE (essential MCU regulator); and the regulatory subunits MICU1-MICU3 (mitochondrial calcium uptake 1-3). Reconstitution studies in yeast, which lacks a mitochondrial Ca2+ uniporter, have demonstrated that heterologous co-expression of MCU and EMRE is necessary and sufficient to confer uniporter activity. The family of EF-hand-containing proteins (MICU1, MICU2, and MICU3) has been shown to exhibit a gatekeeper function for the uniporter, inhibiting Ca2+ uptake at low cytoplasmic concentrations. These components are generally highly conserved across eukaryotes, including most metazoans and plants, but not in many fungi and protozoans, reflecting their ancient and fundamental role (Tufi, 2019).
Although the composition and function of the uniporter have been well characterized in vitro and in cell culture models, the physiological role of the uniporter is beginning to emerge with in vivo characterization of knockout mutants. Current data present a complex picture. Initial studies of MCU knockout mice described a viable strain with a modest phenotype in a mixed genetic background, although subsequent studies using an inbred background reported MCU loss to be lethal or semi-viable and tissue-specific conditional knockout revealed an important role in cardiac homeostasis. Similarly, loss of MICU1 in mice has a complex phenotype, varying from fully penetrant perinatal lethality to incomplete lethality with a range of neuromuscular defects that unexpectedly improve over time in surviving animals (Tufi, 2019).
One explanation for the reported phenotypic variability is that perturbing mitochondrial Ca2+ uptake can be influenced by additional factors, the most obvious being genetic background. Hence, there is a need for greater investigation into the physiological consequences of genetic manipulation of the uniporter components in a genetically powerful model system. This paper reports a comprehensive genetic analysis of the uniporter complex components that are conserved in Drosophila. This includes loss-of-function mutants for MCU, EMRE, MICU1, and MICU3 (Drosophila lack MCUb and MICU2) and corresponding inducible transgenic expression lines. Despite lacking fast Ca2+ uptake, MCU and EMRE mutants present a surprising lack of organismal phenotypes, although both mutants are short lived, with a more pronounced effect when MCU is lost. In contrast, loss of MICU1 causes developmental lethality, whereas mutants for MICU3 are viable with modest phenotypes. Performing genetic interaction studies with these strains, this study has confirmed the gatekeeper function of MICU1 is conserved in flies and reveal that MICU1 and MICU3 are not functionally interchangeable. More surprisingly, it was found that loss of MCU or EMRE does not suppress MICU1 mutant lethality, suggesting that the lethality results from MCU-independent functions. The generation of these genetic tools in Drosophila will facilitate further investigation of the functional roles of the uniporter components in vivo (Tufi, 2019).
MCU mutants are viable and fertile with no gross morphological or behavioral defects, which was initially surprising given the historical importance of mitochondrial Ca2+. Still, this corroborates another report of fly MCU mutants and is consistent with studies in mice and worms in which deletion of MCU orthologs is essentially benign at the organismal level under basal conditions. However, fly MCU mutants are significantly shorter lived than controls. This situation is mirrored by EMRE mutants, albeit with a smaller impact on lifespan. The reason for the shortened lifespans is unknown but may reflect the effects of a chronic bioenergetic deficit evident from the OCR measurements. Accordingly, MCU mutants show a greater respiration defect compared to EMRE mutants, consistent with their respective impacts on lifespan. The respiratory impairment could be due to the previously reported increase in oxidative stress that occurs in MCU mutants, which has yet to be assessed in EMRE mutants. Alternatively, the short lifespan may be due to a myriad of potential metabolic imbalances, such as disruption of NADH/NAD+ levels. Chronic adaptations may also occur through transcriptional responses. Further studies analyzing the metabolic and transcriptional changes occurring in these flies will shed light on this fundamental question (Tufi, 2019).
Nevertheless, the EMRE mutants are relatively benign at the organismal level, which corroborates the surprising viability of MCU mutants. Considering this, it is striking that flies, like mice and worms, consistently show an ability to compensate for the lack of fast mitochondrial Ca2+ uptake, suggesting the induction of some adaptive mechanism. While alternative routes of mitochondrial Ca2+ entry must exist, because matrix Ca2+ is not abolished in MCU knockout (KO) mice, proposed mechanisms are speculative, and it is unclear whether they constitute a compensatory adaptation for fast Ca2+ uptake or simply allow gradual, slow accumulation. However, rapid mitochondrial Ca2+ uptake mediated by MCU is thought to constitute a specific metabolic regulatory mechanism, e.g., to increase ATP production, under certain conditions, such as strenuous exercise or pathological conditions, which is partly evident in the MCU KO mice or heart-specific conditional KO. Such important physiological roles would not necessarily be apparent under basal conditions in flies. MCU has also been proposed to promote wound healing; however, preliminary studies did not find evidence supporting this. The current study presents a summary of the requirements of uniporter components under basal conditions, and further work will be needed to evaluate the role of the uniporter in the full range of physiological conditions (Tufi, 2019).
In seeking to understand the importance of the regulatory components of the uniporter, this study also developed loss-of-function models for MICU1 and MICU3. In contrast to MCU and EMRE mutants, loss of MICU1 results in larval lethality, which is associated with alterations in mitochondrial distribution and motility, and a reduced level of total ATP. In line with its role as the principle gatekeeper of the uniporter, coupled with excess mitochondrial Ca2+ triggering cell death, it was reasoned that the lethality was due to Ca2+ accumulation in the mitochondrial matrix through unregulated MCU-EMRE channels. Supporting this, it was observed that dual overexpression of MCU and EMRE in the eye leads to substantial loss of retinal tissue; concomitant overexpression of MICU1 is sufficient to prevent this phenotype, consistent with MICU1 re-establishing appropriately regulated uniporter channels (Tufi, 2019).
However, one observation that was most surprising was the inability of MCU or EMRE mutants to rescue the MICU1 mutant lethality. This result is particularly puzzling, because it has been shown that mice lacking MICU1, which present multiple pathogenic phenotypes, are substantially rescued by genetic reduction of EMRE levels. While the reason for the lack of rescue in flies is unclear, it is postulate that this suggests the function of MICU1 is not limited to uniporter-dependent Ca2+ uptake. It is not known whether the lethality of MICU1 mutants is specifically due to excessive mitochondrial Ca2+ levels; however, it appears to be independent of fast mitochondrial Ca2+ uptake, because this is eliminated in MCU and EMRE mutants. As noted earlier, other routes of Ca2+ uptake into mitochondria exist, but the mechanisms that regulate them are uncertain. It is possible that aberrant manganese uptake, as reported to occur in cell models, may contribute to the MICU1 mutant lethality. However, this mechanism would presumably be expected to be mitigated by loss of MCU. Nevertheless, these Drosophila models are ideally suited for unbiased genetic screening to uncover such fundamental regulatory mechanisms (Tufi, 2019).
In contrast to MICU1, loss of MICU3 was well tolerated overall at the organismal level. Functional analysis of MICU3 is extremely limited, but the neuronally restricted expression led to the expectation that these mutants might have more neurological-specific phenotypes, which was at least partly borne out. Whereas longevity of these mutants was only minimally affected, they exhibited a notable locomotor deficit even in young flies. It was initially hypothesized that MICU3 may be able to act redundantly with MICU1, but attempts to transgenically rescue MICU1 mutants by ectopic MICU3 expression were unsuccessful. This result is consistent with a report showing that MICU3 binds to MICU1 but apparently enhances mitochondrial Ca2+ uptake (Tufi, 2019).
In summary, this study presents a comprehensive analysis of the conserved components of the mitochondrial Ca2+ importer and its regulators. While loss of the various components results in dramatically different organismal phenotypes, ranging from the most severe deficit exemplified by the MICU1 mutants to the mild consequences of mutating MICU3, such diverse phenotypes mirror the situation reported in humans so far. The first described patients with MICU1 mutations exhibit a severe, complex neurological condition accompanied by muscular dystrophy and congenital myopathy, clearly associated with mitochondrial dysfunction, whereas a later study reported MICU1 patients with a relatively mild fatigue syndrome. One explanation for the reported phenotypic variability is that the consequence of perturbing mitochondrial Ca2+ uptake can be influenced by additional factors, the most obvious being genetic background. The genetic tools described in this study open up the possibility for a thorough analysis of the uniporter function in a powerful genetic model organism, which will advance understanding of the role of mitochondrial Ca2+ in health and disease (Tufi, 2019).
Mitochondria are highly dynamic organelles. Through a large-scale in vivo RNA interference (RNAi) screen that covered around a quarter of the Drosophila melanogaster genes (4000 genes), this study has identified 578 genes whose knockdown led to aberrant shapes or distributions of mitochondria. The complex analysis revealed that knockdown of the subunits of proteasomes, spliceosomes, and the electron transport chain complexes could severely affect mitochondrial morphology. The loss of Dhpr, a gene encoding an enzyme catalyzing tetrahydrobiopterin regeneration, leads to a reduction in the numbers of tyrosine hydroxylase neurons, shorter lifespan, and gradual loss of muscle integrity and climbing ability. The affected mitochondria in Dhpr mutants are swollen and have fewer cristae, probably due to lower levels of Drp1 S-nitrosylation. Overexpression of Drp1, but not of S-nitrosylation-defective Drp1, rescued Dhpr RNAi-induced mitochondrial defects. It is proposed that Dhpr regulates mitochondrial morphology and tissue homeostasis by modulating S-nitrosylation of Drp1 (Zhou, 2019).
Mitochondria are central to both energy metabolism and biosynthesis. Mitochondrial function could therefore influence resource allocation. Critically, mitochondrial function depends on interactions between proteins encoded by the mitochondrial and nuclear genomes. Severe incompatibilities between these genomes can have pervasive effects on both fitness and longevity. How milder deficits in mitochondrial function affect life-history trade-offs is less well understood. This study analysed how mitonuclear interactions affect the trade-off between fecundity and longevity in Drosophila melanogaster. A panel of 10 different mitochondrial DNA haplotypes are considered against two contrasting nuclear backgrounds (w(1118) (WE) and Zim53 (ZIM)) in response to high-protein versus standard diet.Strikingly different responses are reported between the two nuclear backgrounds. WE females have higher fecundity and decreased longevity on high protein. ZIM females have much greater fecundity and shorter lifespan than WE flies on standard diet. High protein doubled their fecundity with no effect on longevity. Mitochondrial haplotype reflected nuclear life-history trade-offs, with a negative correlation between longevity and fecundity in WE flies and no correlation in ZIM flies. Mitonuclear interactions had substantial effects but did not reflect genetic distance between mitochondrial haplotypes. It is concluded that mitonuclear interactions can have significant impact on life-history trade-offs, but their effects are not predictable by relatedness. This article is part of the theme issue 'Linking the mitochondrial genotype to phenotype: a complex endeavour' (Camus, 2020)
A mutant mitochondrial genome arising amid the pool of mitochondrial genomes within a cell must compete with existing genomes to survive to the next generation. Even weak selective forces can bias transmission of one genome over another to affect the inheritance of mitochondrial diseases and guide the evolution of mitochondrial DNA (mtDNA). Studies in several systems suggested that purifying selection in the female germline reduces transmission of detrimental mitochondrial mutations. In contrast, some selfish genomes can take over despite a cost to host fitness. Within individuals, the outcome of competition is therefore influenced by multiple selective forces. The nuclear genome, which encodes most proteins within mitochondria, and all external regulators of mitochondrial biogenesis and dynamics can influence the competition between mitochondrial genomes, yet little is known about how this works. Previous work established a Drosophila line transmitting two mitochondrial genomes in a stable ratio enforced by purifying selection benefiting one genome and a selfish advantage favoring the other. In this study, to find nuclear genes that impact mtDNA competition, heterozygous deletions were screened tiling approximately 70% of the euchromatic regions, and their influence on this ratio was examined. This genome-wide screen detected many nuclear modifiers of this ratio and identified one as the catalytic subunit of mtDNA polymerase gene (POLG), tam. A reduced dose of tam drove elimination of defective mitochondrial genomes. This study suggests that this approach will uncover targets for interventions that would block propagation of pathogenic mitochondrial mutations (Chiang, 2019).
Translating advances in cancer research to clinical applications requires better insight into the metabolism of normal cells and tumour cells in vivo. Much effort has focused on understanding how glycolysis and oxidative phosphorylation (OxPhos) support proliferation, while their impact on other aspects of development and tumourigenesis remain largely unexplored. This study found that inhibition of OxPhos in neural stem cells (NSCs) or tumours in the Drosophila brain not only decreases proliferation, but also affects many different aspects of stem cell behaviour. In NSCs, OxPhos dysfunction leads to a protracted G1/S-phase and results in delayed temporal patterning and reduced neuronal diversity. As a consequence, NSCs fail to undergo terminal differentiation, leading to prolonged neurogenesis into adulthood. Similarly, in brain tumours inhibition of OxPhos slows proliferation and prevents differentiation, resulting in reduced tumour heterogeneity. Thus, in vivo, highly proliferative stem cells and tumour cells require OxPhos for efficient growth and generation of diversity (van den Ameele, 2019).
The observation that some cancer cells rely primarily on aerobic glycolysis for energy and biomass production (the Warburg effect) has often led to the assumption that the other main source of ATP, mitochondrial oxidative phosphorylation (OxPhos), is dispensable. However, it is becoming increasingly clear that many tumours do require mitochondrial activity for energy and biosynthesis and OxPhos is now frequently exploited as a therapeutic target in cancer. OxPhos takes place at the inner mitochondrial membrane in five large protein complexes (Complex I-V), which together form the respiratory chain. Complexes I-IV transfer electrons from NADH to O2 and use the released energy to translocate protons from the mitochondrial matrix into the intermembrane space. The resulting electrochemical gradient is then used by Complex V (ATP synthase) to generate ATP from ADP. Apart from the production of ATP, OxPhos is also directly involved in the generation of NAD+, orotate, fumarate and reactive oxygen species (ROS) and thus affects many cellular processes, such as nucleotide synthesis, signalling pathway activity and epigenetic modifications. The Warburg effect has since been interpreted as a normal adaptation to the metabolic requirements of proliferation, both in cancer cells and proliferating stem cells. High glycolytic flux is thought to be required for a constant supply of biomass while OxPhos, apart from its role in production of ATP, primarily maintains the cellular redox balance (van den Ameele, 2019 and references therein).
However, metabolic flux in cancer cells can be influenced by extrinsic and intrinsic factors such as substrate availability, oncogenic mutations and the tumour's tissue and cell type of origin. Brain tumours in particular recapitulate many features of their tissue of origin and grow along a hierarchy reminiscent of normal brain development. An integrated understanding of the interactions between metabolism and cell identity in vivo, during both tumourigenesis and normal development, is therefore crucial to translate advances in cancer research to clinical applications (van den Ameele, 2019).
Development of the Drosophila central nervous system (CNS) has been used extensively as a powerful reductionist model of human brain development and tumourigenesis in vivo. The CNS of Drosophila develops from rapidly cycling embryonic and larval neural stem cells (NSCs) that generate a wide variety of neurons and glia. Neuronal diversity is achieved primarily by spatial and temporal patterning, which confers specific identities on NSCs and their progeny according to their location and developmental time. Neural stem cells (NSCs) in Drosophila and mammals are thought to generate ATP through aerobic glycolysis rather than OxPhos, whereas their neuronal progeny switch to mitochondrial respiration upon differentiation. Upregulation of aerobic glycolysis, reminiscent of the Warburg effect, has also been described in a number of Drosophila tumour paradigms. However, the interpretation that mitochondrial respiration is dispensable for normal Drosophila NSCs contrasts with the clear requirement for OxPhos to support cell cycle progression in the Drosophila eye disc. This study investigated whether, and to what extent, Drosophila NSCs and brain tumours rely on oxidative phosphorylation (van den Ameele, 2019).
This study shows that the metabolic requirements of highly proliferative NSCs in the Drosophila brain, as well as the tumour cells NSCs generate upon transformation, cannot be met by aerobic glycolysis alone. Instead, Drosophila NSCs require OxPhos for key aspects of their behaviour: proliferation, generation of diversity through temporal patterning, and termination of proliferation. Respiratory activity may provide an explanation for the strong increase in ROS production that has been observed in NSCs upon hypoxia and for the developmental lethality caused by CNS-specific mutation of the mitochondrial genome. While OxPhos dysfunction affects both normal NSCs and tumour cells in the brain, inhibition of glycolysis only affects tumour growth but not normal brain development. This is reminiscent of the upregulation of aerobic glycolysis in Hipk, EGFR or PDGF/VEGF-induced tumours in the Drosophila wing disc. Future experiments will determine the origin and consequences of this tumour-specific reliance on glycolysis in the brain (van den Ameele, 2019).
The results contrast with previous findings suggesting that OxPhos is dispensable during normal NSC development and in brain tumours, and is only activated at the end of neurogenesis as part of a metabolic switch to induce termination of NSC proliferation. While the current experiments do not directly address whether this metabolic switch takes place, the results provide an alternate interpretation. Sustained OxPhos activity throughout NSC development was shown to be required for normal temporal patterning. Prolonged expression of early temporal markers makes NSCs unresponsive to the developmental cues that govern cell cycle exit and this study shows that restoring temporal progression by timely depletion of the early temporal factor Imp enhances termination of proliferation in spite of continued OxPhos inhibition. These findings thus integrate key aspects of NSC and tumour cell biology: OxPhos-dependent proliferation is required for temporal patterning and differentiation at the G1/S transition of the cell cycle. This enables NSCs to undergo normal aging and to respond to the developmental cues that instruct termination of proliferation. Interestingly, adult neurogenesis in the subventricular zone of the mammalian brain depends on p57-induced slowing of the cell cycle during embryonic development. It is not known whether p57 expression or mitochondrial dysfunction also affects the temporal identity of mammalian NSCs. Importantly, the effects observed in this study are specific to the G1/S transition: activation of the G2/M checkpoint did not affect temporal patterning or termination of proliferation. The results therefore demonstrate that the size and composition of Drosophila NSC lineages are not strictly predetermined but rather controlled by both intrinsic and extrinsic factors. Single-cell sequencing data indicate that metabolic differences exist between NSCs in different regions of the brain or at different developmental stages, and it will be interesting to assess whether all NSCs are similarly affected by OxPhos dysfunction and G1/S delay or whether specific lineages show stereotypical responses, as has been shown for entry into quiescence, where arrest in G2 or G0 is predetermined (van den Ameele, 2019).
This study indicates that OxPhos might constitute a targetable metabolic vulnerability of cancer. Small molecule inhibitors of OxPhos are currently being developed and tested in clinical trials to treat various forms of cancer. However, this study found that the in vivo impact of OxPhos dysfunction is much more complex than mere inhibition of proliferation. A better understanding of the interactions between metabolism, differentiation and tumour heterogeneity in vivo has the potential to uncover novel therapeutic approaches (van den Ameele, 2019).
Metabolic reprogramming is a key feature of many cancers, but how and when it contributes to tumorigenesis remains unclear. This study demonstrates that metabolic reprogramming induced by mitochondrial fusion can be rate-limiting for immortalization of tumor-initiating cells (TICs) and trigger their irreversible dedication to tumorigenesis. Using single-cell transcriptomics, this study found that Drosophila brain tumors contain a rapidly dividing stem cell population defined by upregulation of oxidative phosphorylation (OxPhos). Targeted metabolomics and in vivo genetic screening were combined to demonstrate that OxPhos is required for tumor cell immortalization but dispensable in neural stem cells (NSCs) giving rise to tumors. Employing an in vivo NADH/NAD(+) sensor, it was shown that NSCs precisely increase OxPhos during immortalization. Blocking OxPhos or mitochondrial fusion stalls TICs in quiescence and prevents tumorigenesis through impaired NAD(+) regeneration. This work establishes a unique connection between cellular metabolism and immortalization of tumor-initiating cells (Bonnay, 2020).
Organismal fitness is partly determined by how well the nutritional intake matches sex-specific metabolic requirements. Metabolism itself is underpinned by complex genomic interactions involving products from both nuclear and mitochondrial genomes. Products from these two genomes must coordinate how nutrients are extracted, used and recycled, processes vital for fuelling reproduction. Given the complicated nature of metabolism, it is not well understood how the functioning of these two genomes is modulated by nutrients. This study used nutritional geometry techniques on Drosophila lines that only differ in their mtDNA, with the aim to understand if there is nutrient-dependent mitochondrial genetic variance for male reproduction. First, genetic variance was found for diet consumption, indicating that flies are consuming different amounts of food to meet new physiological requirements. Then an interaction was found between mtDNA and diet for fitness, suggesting that the mtDNA plays a role in modulating diet-dependent fitness. These results enhance basic understanding of nutritional health and of chimeric genomes (Camus, 2020).
Interactions between cytoplasmic and nuclear genomes confer sex-specific effects on lifespan in Drosophila melanogaster
Genetic variation outside of the cell nucleus can affect the phenotype. The cytoplasm is home to the mitochondria, and in arthropods often hosts intracellular bacteria such as Wolbachia. While numerous studies have implicated epistatic interactions between cytoplasmic and nuclear genetic variation as mediators of phenotypic expression, two questions remain. Firstly, it remains unclear whether outcomes of cyto-nuclear interactions will manifest differently across the sexes, as might be predicted given that cytoplasmic genomes are screened by natural selection only through females as a consequence of their maternal inheritance. Secondly, the relative contribution of mitochondrial genetic variation to other cytoplasmic sources of variation, such as Wolbachia infection, in shaping phenotypic outcomes of cyto-nuclear interactions remains unknown. This study addresses these questions, creating a fully-crossed set of replicated cyto-nuclear populations derived from three geographically distinct populations of Drosophila melanogaster, measuring the lifespan of males and females from each population. It was observed that cyto-nuclear interactions shape lifespan, and that the outcomes of these interactions differ across the sexes. Yet, no evidence was found that placing the cytoplasms from one population alongside the nuclear background of others (generating putative cyto-nuclear mismatches) leads to decreased lifespan in either sex. Although it was difficult to partition mitochondrial from Wolbachia effects, the results suggest at least some of the cytoplasmic genotypic contribution to lifespan was directly mediated by an effect of sequence variation in the mtDNA. Future work should explore the degree to which cyto-nuclear interactions result in sex differences in expression of other components of organismal life-history (Vaught, 2020).
Friedreich ataxia (FRDA) is a neurodegenerative disorder characterized by neuromuscular and neurological manifestations. It is caused by mutations in the FXN gene, which results in loss of the mitochondrial protein frataxin. Endoplasmic Reticulum-mitochondria associated membranes (MAMs) are inter-organelle structures involved in the regulation of essential cellular processes, including lipid metabolism and calcium signaling. This study has analyzed in both, unicellular and multicellular models of FRDA, calcium management and integrity of MAMs. Function of MAMs was observed to be compromised in the cellular model of FRDA, which was improved upon treatment with antioxidants. In agreement, promoting mitochondrial calcium uptake was sufficient to restore several defects caused by frataxin deficiency in Drosophila melanogaster. Remarkably, the findings describe for the first time frataxin as a member of the protein network of MAMs, where interacts with two of the main proteins implicated in endoplasmic reticulum-mitochondria communication. These results suggest a new role of frataxin, indicate that FRDA goes beyond mitochondrial defects and highlight MAMs as novel therapeutic candidates to improve patient's conditions (Rodríguez, 2020).
Precise regulation of stem cell activity is crucial for tissue homeostasis. In Drosophila, intestinal stem cells (ISCs) maintain the midgut epithelium and respond to oxidative challenges. However, the connection between intestinal homeostasis and redox signaling remains obscure. This study found that Caliban (Clbn), a component of the ribosome quality control complex (RQC), functions as a regulator of mitochondrial dynamics in enterocytes (ECs) and is required for intestinal homeostasis. The clbn knock-out flies have a shortened lifespan and lose the intestinal homeostasis. Clbn is highly expressed and localizes to the outer membrane of mitochondria in ECs. Mechanically, Clbn mediates mitochondrial dynamics in ECs and removal of clbn leads to mitochondrial fragmentation, accumulation of reactive oxygen species, ECs damage, activation of JNK and JAK-STAT signaling pathways. Moreover, multiple mitochondria-related genes are differentially expressed between wild-type and clbn mutated flies by a whole-genome transcriptional profiling. Furthermore, loss of clbn promotes tumor growth in gut generated by activated Ras in intestinal progenitor cells. These findings reveal an EC-specific function of Clbn in regulating mitochondrial dynamics, and provide new insight into the functional link among mitochondrial redox modulation, tissue homeostasis and longevity (Cai, 2020).
Although mitochondrial dysfunction is associated with the development and progression of diabetic nephropathy (DN), its mechanisms are poorly understood, and it remains debatable whether mitochondrial morphological change is a cause of DN. In this study, a Drosophila DN model was established by treating a chronic high-sucrose diet that exhibits similar phenotypes in animals. Results showed that flies fed a chronic high-sucrose diet exhibited a reduction in lifespan, as well as increased lipid droplets in fat body tissue. Furthermore, the chronic high-sucrose diet effectively induced the morphological abnormalities of nephrocytes in Drosophila. High-sucrose diet induced mitochondria fusion in nephrocytes by increasing Opa1 and Marf expression. These findings establish Drosophila as a useful model for studying novel regulators and molecular mechanisms for imbalanced mitochondrial dynamics in the pathogenesis of DN. Furthermore, understanding the pathology of mitochondrial dysfunction regarding morphological changes in DN would facilitate the development of novel therapeutics (Kim, 2021).
Traumatic brain injuries (TBIs) caused by a sudden impact to the head alter behavior and impair physical and cognitive function. Besides the severity, type and area of the brain affected, the outcome of TBI is also influenced by the patient's biological sex. Previous studies reporting mitochondrial dysfunction mainly focused on exponential reactive oxygen species (ROS) generation, increased mitochondrial membrane potential, and altered mitochondrial dynamics as a key player in the outcome to brain injury. This study evaluated the effect of a near-infrared (NIR) light exposure on gene expression in a Drosophila TBI model. NIR interacts with cytochrome c oxidase (COX) of the electron transport chain to reduce mitochondrial membrane potential hyperpolarization, attenuate ROS generation, and apoptosis. W (1118) male and female flies were subjected to TBI using a high-impact trauma (HIT) device and subsequently exposed the isolated fly brains to a COX-inhibitory wavelength of 750 nm for 2 hours (hr). Genome-wide 3'-mRNA-sequencing of fly brains revealed that injured w (1118) females exhibit greater changes in transcription compared to males at 1, 2, and 4 hours (hr) after TBI. Inhibiting COX by exposure to NIR downregulates gene expression in injured females but has minimal effect in injured males. These results suggest that mitochondrial COX modulation with NIR alters gene expression in Drosophila following TBI and the response to injury and NIR exposure varies by biological sex (Shah, 2021).
Eukaryotic cells maintain proteostasis, the balance between protein synthesis, folding, re-folding and degradation, through mechanisms that require cytoplasmic and mitochondrial translation. Genetic defects affecting cytoplasmic translation perturb synapse development, neurotransmission, and are causative of neurodevelopmental disorders such as Fragile X syndrome. In contrast, there is little indication that mitochondrial proteostasis, either in the form of mitochondrial protein translation and/or degradation, is required for synapse development and function. This study focused on two genes deleted in a recurrent copy number variation causing neurodevelopmental disorders, the 22q11.2 microdeletion syndrome. SLC25A1 and MRPL40, two genes present in the microdeleted segment and whose products localize to mitochondria, interact and are necessary for mitochondrial ribosomal integrity and proteostasis. Drosophila studies show that mitochondrial ribosome function is necessary for synapse neurodevelopment, function, and behavior. It is proposed that mitochondrial proteostasis perturbations, either by genetic or environmental factors, are a pathogenic mechanism for neurodevelopmental disorders (Gokhale, 2021).
In addition to their well characterized role in cellular energy production, new evidence has revealed the involvement of mitochondria in diverse signaling pathways that regulate a broad array of cellular functions. The mitochondrial genome (mtDNA) encodes essential components of the oxidative phosphorylation (OXPHOS) pathway whose expression must be coordinated with the components transcribed from the nuclear genome. Mitochondrial dysfunction is associated with disorders including cancer and neurodegenerative diseases, yet the role of the complex interactions between the mitochondrial and nuclear genomes are poorly understood. Using a Drosophila model in which alternative mtDNAs are present on a common nuclear background, the effects of this altered mitonuclear communication on the transcriptomic response to altered nutrient status were studied. Adult flies with the 'native' and 'disrupted' genotypes were re-fed following brief starvation, with or without exposure to rapamycin, the cognate inhibitor of the nutrient-sensing target of rapamycin (TOR). RNAseq showed that alternative mtDNA genotypes affect the temporal transcriptional response to nutrients in a rapamycin-dependent manner. Pathways most greatly affected were OXPHOS, protein metabolism and fatty acid metabolism. A distinct set of testis-specific genes was also differentially regulated in the experiment. It is concluded that any of the differentially expressed genes between alternative mitonuclear genotypes have no direct interaction with mtDNA gene products, suggesting that the mtDNA genotype contributes to retrograde signaling from mitochondria to the nucleus. The interaction of mitochondrial genotype (mtDNA) with rapamycin treatment identifies new links between mitochondria and the nutrient-sensing mTORC1 (mechanistic target of rapamycin complex 1) signaling pathway (Santiago, 2021).
The balanced functionality of cellular proteostatic modules is central to both proteome stability and mitochondrial physiology; thus, the age-related decline of proteostasis also triggers mitochondrial dysfunction, which marks multiple degenerative disorders. Non-functional mitochondria are removed by mitophagy, including Parkin/Pink1-mediated mitophagy. A common feature of neuronal or muscle degenerative diseases, is the accumulation of damaged mitochondria due to disrupted mitophagy rates. This study exploited Drosophila as a model organism to investigate the functional role of Parkin/Pink1 in regulating mitophagy and proteostatic responses, as well as in suppressing degenerative phenotypes at the whole organism level. Parkin or Pink1 knock down in young flies modulated proteostatic components in a tissue-dependent manner, increased cell oxidative load, and suppressed mitophagy in neuronal and muscle tissues, causing mitochondrial aggregation and neuromuscular degeneration. Concomitant to Parkin or Pink1 knock down cncC/Nrf2 overexpression, induced the proteostasis network, suppressed oxidative stress, restored mitochondrial function, and elevated mitophagy rates in flies' tissues; it also, largely rescued Parkin or Pink1 knock down-mediated neuromuscular degenerative phenotypes. These in vivo findings highlight the critical role of the Parkin/Pink1 pathway in mitophagy, and support the therapeutic potency of Nrf2 (a druggable pathway) activation in age-related degenerative diseases (Gumeni, 2021).
Parkinson's disease (PD) is a progressive neurodegenerative disorder with no known cure. PD is characterized by locomotion deficits, nigrostriatal dopaminergic neuronal loss, mitochondrial dysfunctions and formation of α-Synuclein aggregates. A well-conserved and less understood family of Tubulin Polymerization Promoting Proteins (TPPP) is also implicated in PD and related disorders, where TPPP exists in pathological aggregates in neurons in patient brains. However, there are no in vivo studies on mammalian TPPP to understand the genetics and neuropathology linking TPPP aggregation or neurotoxicity to PD. The only Drosophila homolog of human TPPP is named Ringmaker (Ringer). This study reports that adult ringer mutants display progressive locomotor disabilities, reduced lifespan and neurodegeneration. Importantly, the findings reveal that Ringer is associated with mitochondria and ringer mutants have mitochondrial structural damage and dysfunctions. Adult ringer mutants also display progressive loss of dopaminergic neurons. Together, these phenotypes of ringer mutants recapitulate some of the salient features of human PD patients, thus allowing utilization of ringer mutants as a fly model relevant to PD, and further exploration of its genetic and molecular underpinnings to gain insights into the role of human TPPP in PD (Xie, 2021).
Mitochondrial DNA (mtDNA) encodes gene products that are essential for oxidative phosphorylation. They organize as higher order nucleoid structures (mtNucleoids) that were shown to be critical for the maintenance of mtDNA stability and integrity. While mtNucleoid structures are associated with cellular health, how they change in situ under physiological maturation and aging requires further investigation. This study investigated the mtNucleoid assembly at an ultrastructural level in situ using the TFAM-Apex2 Drosophila model. Smaller and more compact TFAM-nucleoids are populated in the mitochondria of indirect flight muscle of aged flies. Furthermore, mtDNA transcription and replication were cross-regulated in the mtTFB2-knockdown flies as in the mtRNAPol-knockdown flies that resulted in reductions in mtDNA copy numbers and nucleoid-associated TFAM. Overall, this study reveals that the modulation of TFAM-nucleoid structure under physiological aging, which is critically regulated by mtDNA content (Wang, 2021).
Various neurodegenerative disorders are associated with human NTE/PNPLA6 dysfunction. Mechanisms of neuropathogenesis in these diseases are far from clearly elucidated. Hereditary spastic paraplegia belongs to a type of neurodegeneration associated with NTE/PNLPLA6 and is implicated in neuron death. This study used Drosophila melanogaster to investigate the consequences of neuronal knockdown of swiss cheese (sws)-the evolutionarily conserved ortholog of human NTE/PNPLA6-in vivo. Adult flies with the knockdown show longevity decline, locomotor and memory deficits, severe neurodegeneration progression in the brain, reactive oxygen species level acceleration, mitochondria abnormalities and lipid droplet accumulation. These results suggest that SWS/NTE/PNPLA6 dysfunction in neurons induces oxidative stress and lipid metabolism alterations, involving mitochondria dynamics and lipid droplet turnover in neurodegeneration pathogenesis. It is proposed that there is a complex mechanism in neurological diseases such as hereditary spastic paraplegia, which includes a stress reaction, engaging mitochondria, lipid droplets and endoplasmic reticulum interplay (Melentev, 2021).
Neurodegenerative disease (ND) is a growing health burden worldwide, but its causes and treatments remain elusive. Although most cases of ND are sporadic, rare familial cases have been attributed to single genes, which can be investigated in animal models. This study generated a new mutation in the calcium-independent phospholipase A2 (iPLA2) VIA gene CG6718, the Drosophila melanogaster ortholog of human PLA2G6/PARK14, mutations in which cause a suite of NDs collectively called PLA2G6-associated neurodegeneration (PLAN). The mutants display age-related loss of climbing ability, a symptom of neurodegeneration in flies. Although phospholipase activity commonly is presumed to underlie iPLA2-VIA function, locomotor decline in the mutants used in this study is rescued by a transgene carrying a serine-to-alanine mutation in the catalytic residue, suggesting that important functional aspects are independent of phospholipase activity. Additionally, it was found that iPLA2-VIA knockdown in either muscle or neurons phenocopies locomotor decline with age, demonstrating its necessity in both neuronal and non-neuronal tissues. Furthermore, RNA in situ hybridization shows high endogenous iPLA2-VIA mRNA expression in adult germ cells, and transgenic HA-tagged iPLA2-VIA colocalizes with mitochondria there. Mutant males are fertile with normal spermatogenesis, while fertility is reduced in mutant females. Mutant female germ cells display age-related mitochondrial aggregation, loss of mitochondrial potential, and elevated cell death. These results suggest that iPLA2-VIA is critical for mitochondrial integrity in the Drosophila female germline, which may provide a novel context to investigate its functions with parallels to PLAN (Banerjee, 2021).
Individual hosts within populations often show inter-individual variation in their susceptibility to bacterial pathogen-related diseases. Utilizing Drosophila, this study highlighted that phenotypic variation in host-pathogen susceptibility within populations is driven by energetic trade-offs, facilitated by infection-mediated changes in glutamate metabolism. Furthermore, host-pathogen susceptibility is conditioned by life history, which adjusts immunometabolic sensing in muscles to direct vitamin-dependent reallocation of host energy substrates from the adipose tissue (i.e., a muscle-adipose tissue axis). Life history conditions inter-individual variation in the activation strength of intra-muscular NF-κB signaling. Limited intra-muscular NF-κB signaling activity allows for enhanced infection-mediated mitochondrial biogenesis and function, which stimulates glutamate dehydrogenase-dependent synthesis of glutamate. Muscle-derived glutamate acts as a systemic metabolite to promote lipid mobilization through modulating vitamin B enzymatic cofactor transport and function in the adipose tissue. This energy substrate reallocation improves pathogen clearance and boosts host survival. Finally, life history events that adjust energetic trade-offs can shape inter-individual variation in host-pathogen susceptibility after infection (Zhao, 2021).
Cardiolipin (CL) deficiency causes mitochondrial dysfunction and aberrant metabolism that are associated in humans with the severe disease Barth syndrome (BTHS). Several metabolic abnormalities are observed in BTHS patients and model systems, including decreased oxidative phosphorylation, reduced tricarboxylic acid (TCA) cycle flux, and accumulated lactate and D-β-hydroxybutyrate, which strongly suggests that nicotinamide adenine dinucleotide (NAD) redox metabolism may be altered in CL-deficient cells. This study identified abnormal NAD(+) metabolism in multiple BTHS model systems and demonstrate that supplementation of NAD(+) precursors such as nicotinamide mononucleotide (NMN) improves mitochondrial function. Improved mitochondrial function in the Drosophila model was associated with restored exercise endurance, which suggests a potential therapeutic benefit of NAD(+) precursor supplementation in the management of BTHS patients (Ji, 2022).
Age-related memory impairment (AMI) occurs in many species, including humans. The underlying mechanisms are not fully understood. In wild-type Drosophila (w1118), AMI appears in the form of a decrease in learning (3-min memory) from middle age (30 days after eclosion [DAE]). in vivo, DNA microarray, and behavioral screen studies were performed to identify genes controlling both lifespan and AMI and mitochondrial Acon1 (mAcon1) was selected. mAcon1 expression in the head of w(1118) decreased with age. Neuronal overexpression of mAcon1 extended its lifespan and improved AMI. Neuronal or mushroom body expression of mAcon1 regulated the learning of young (10 DAE) and middle-aged flies. Interestingly, acetyl-CoA and citrate levels increased in the heads of middle-aged and neuronal mAcon1 knockdown flies. Acetyl-CoA, as a cellular energy sensor, is related to autophagy. Autophagy activity and efficacy determined by the positive and negative changes in the expression levels of Atg8a-II and p62 were proportional to the expression level of mAcon1. Levels of the presynaptic active zone scaffold protein Bruchpilot were inversely proportional to neuronal mAcon1 levels in the whole brain. Furthermore, mAcon1 overexpression in Kenyon cells induced mitophagy labeled with mt-Keima and improved learning ability. Both processes were blocked by pink1 knockdown. Taken together, these results imply that the regulation of learning and AMI by mAcon1 occurs via autophagy/mitophagy-mediated neural plasticity (Cho, 2021).
Synapses are particularly susceptible to the effects of advancing age, and mitochondria have long been implicated as organelles contributing to this compartmental vulnerability. Despite this, the mitochondrial molecular cascades promoting age-dependent synaptic demise remain to be elucidated. This study sought to examine how the synaptic mitochondrial proteome (including strongly mitochondrial associated proteins) was dynamically and temporally regulated throughout ageing to determine whether alterations in the expression of individual candidates can influence synaptic stability/morphology. Proteomic profiling of wild-type mouse cortical synaptic and non-synaptic mitochondria across the lifespan revealed significant age-dependent heterogeneity between mitochondrial subpopulations, with aged organelles exhibiting unique protein expression profiles. Recapitulation of aged synaptic mitochondrial protein expression at the Drosophila neuromuscular junction has the propensity to perturb the synaptic architecture, demonstrating that temporal regulation of the mitochondrial proteome may directly modulate the stability of the synapse in vivo (Graham, 2021).
Chemotherapy-induced peripheral neuropathy (CIPN) is a prevalent side effect of widely used platinum-based anti-cancer agents. There are few predictable risk factors to identify susceptible patients. Effective preventive measures or treatments are not available. This study used a model of CIPN in Drosophila melanogaster to identify genetic changes that confer resistance to cisplatin-induced neuronal damage but not in the rapidly dividing cells of the ovary. The Drosophila strain attP40, used as a genetic background for creation of RNAi lines, is resistant to cisplatin damage compared to the similar attP2 background strain. attP40 flies have reduced mRNA expression of ND-13A, a component of the mitochondria electron transport chain complex I. Reduction of ND-13A via neuron-specific RNAi leads to resistance to the dose-dependent climbing deficiencies and neuronal apoptosis observed in control flies. These flies are also resistant to acute oxidative stress, suggesting a mechanism for resistance to cisplatin. The mitochondria of attP40 flies function similarly to control attP2 mitochondria under normal conditions. Mitochondria are damaged by cisplatin, leading to reduced activity, but attP40 mitochondria are able to retain function and even increase basal respiration rates in response to this stress. This retained mitochondrial activity is likely mediated by Sirt1 and PGC1α, and is key to cisplatin resistance. These findings represent potential for both identification of susceptible patients and prevention of CIPN through the targeting of mitochondria (Groen, 2021).
Experimental evolution with Drosophila melanogaster has been used extensively for decades to study aging and longevity. In recent years, the addition of DNA and RNA sequencing to this framework has allowed researchers to leverage the statistical power inherent to experimental evolution to study the genetic basis of longevity itself. Here, we incorporated metabolomic data into to this framework to generate even deeper insights into the physiological and genetic mechanisms underlying longevity differences in three groups of experimentally evolved D. melanogaster populations with different aging and longevity patterns. Metabolomic analysis found that aging alters mitochondrial metabolism through increased consumption of NAD(+) and increased usage of the TCA cycle. Combining the genomic and metabolomic data produced a list of biologically relevant candidate genes. Among these candidates, significant enrichment was found for genes and pathways associated with neurological development and function, and carbohydrate metabolism. While enrichment for aging canonical genes was not specifically found, neurological dysregulation and carbohydrate metabolism are both known to be associated with accelerated aging and reduced longevity. Taken together, these results provide plausible genetic mechanisms for what might be driving longevity differences in this experimental system. More broadly, these findings demonstrate the value of combining multiple types of omic data with experimental evolution when attempting to dissect mechanisms underlying complex and highly polygenic traits such as aging (Phillips, 2022).
alpha-Synuclein (α-syn) is important in synucleinopathies such as Parkinson's disease (PD). While genome-wide association studies (GWASs) of synucleinopathies have identified many risk loci, the underlying genes have not been shown for most loci. Using Drosophila, 3,471 mutant chromosomes were screened for genetic modifiers of α-synuclein and 12 genes were identified. Eleven modifiers have human orthologs associated with diseases, including MED13 and CDC27, which lie within PD GWAS loci. Drosophila Skd/Med13 and glycolytic enzymes are co-upregulated by α-syn-associated neurodegeneration. While elevated α-syn compromises mitochondrial function, co-expressing skd/Med13 RNAi and α-syn synergistically increase the ratio of oxidized-to-reduced glutathione. The resulting neurodegeneration can be suppressed by overexpressing a glycolytic enzyme or treatment with deferoxamine, suggesting that compensatory glycolysis is neuroprotective. In addition, the functional relationship between α-synuclein, MED13, and glycolytic enzymes is conserved between flies and mice. It is proposed that hypoxia-inducible factor and MED13 are part of a druggable pathway for PD (Ren, 2023).
The protein DJ-1 is mutated in rare familial forms of recessive Parkinson's disease and in parkinsonism accompanied by amyotrophic lateral sclerosis symptoms and dementia. DJ-1 is considered a multitasking protein able to confer protection under various conditions of stress. However, the precise cellular function still remains elusive. In the present work, fruit flies lacking the expression of the DJ-1 homolog dj-1β were assessed as compared to control aged-matched individuals. Behavioral evaluations included lifespan, locomotion in an open field arena, sensitivity to oxidative insults, and resistance to starvation. Molecular analyses were carried out by analyzing the mitochondrial morphology and functionality, and the autophagic response. It was demonstrated that dj-1β null mutant flies are hypoactive and display higher sensitivity to oxidative insults and food deprivation. Analysis of mitochondrial homeostasis revealed that loss of dj-1β leads to larger and more circular mitochondria, characterized by impaired complex-I-linked respiration while preserving ATP production capacity. Additionally, dj-1β null mutant flies present an impaired autophagic response, which is suppressed by treatment with the antioxidant molecule N-Acetyl-L-Cysteine. Overall, these data point to a mechanism whereby DJ-1 plays a critical role in the maintenance of energy homeostasis, by sustaining mitochondrial homeostasis and affecting the autophagic flux through the maintenance of the cellular redox state. In light of the involvement of DJ-1 in neurodegenerative diseases and considering that neurons are highly energy-demanding cells, particularly sensitive to redox stress, this study sheds light on a key role of DJ-1 in the maintenance of cellular homeostasis (De Lazzari, 2023).
Neuronal intranuclear inclusion disease (NIID) is a neuromuscular/neurodegenerative disease caused by the expansion of CGG repeats in the 5' untranslated region (UTR) of the NOTCH2NLC gene. These repeats can be translated into a polyglycine-containing protein, uN2CpolyG, which forms protein inclusions and is toxic in cell models, albeit through an unknown mechanism. This study established a transgenic Drosophila model expressing uN2CpolyG in multiple systems, which resulted in progressive neuronal cell loss, locomotor deficiency, and shortened lifespan. Interestingly, electron microscopy revealed mitochondrial swelling both in transgenic flies and in muscle biopsies of individuals with NIID. Immunofluorescence and immunoelectron microscopy showed colocalization of uN2CpolyG with mitochondria in cell and patient samples, while biochemical analysis revealed that uN2CpolyG interacted with a mitochondrial RNA binding protein, LRPPRC (leucine-rich pentatricopeptide repeat motif-containing protein). Furthermore, RNA sequencing (RNA-seq) analysis and functional assays showed down-regulated mitochondrial oxidative phosphorylation in uN2CpolyG-expressing flies and NIID muscle biopsies. Finally, idebenone treatment restored mitochondrial function and alleviated neurodegenerative phenotypes in transgenic flies. Overall, these results indicate that transgenic flies expressing uN2CpolyG recapitulate key features of NIID and that reversing mitochondrial dysfunction might provide a potential therapeutic approach for this disorder (Yu, 2023).
Mutations in the Mpv17 gene are responsible for MPV17-related hepatocerebral mitochondrial DNA depletion syndrome and Charcot-Marie-Tooth (CMT) disease. CG11077 (Drosophila Mpv17; dMpv17), an ortholog of human MPV17, was knocked down in the nervous system in Drosophila melanogaster and the behavioral and cellular phenotypes were investigated. The resulting dMpv17 knockdown larvae showed impaired locomotor activity and learning ability consistent with mitochondrial defects suggested by the reductions in mitochondrial DNA and ATP production and the increases in the levels of lactate and reactive oxygen species. Furthermore, an abnormal morphology of the neuromuscular junction, at the presynaptic terminal, was observed in dMpv17 knockdown larvae. These results reproduce well the symptoms of human diseases and partially reproduce the phenotypes of Mpv17-deficient model organisms. Therefore, it is suggested that neuron-specific dMpv17 knockdown in Drosophila is a useful model for investigation of MPV17-related hepatocerebral mitochondrial DNA depletion syndrome and CMT caused by Mpv17 dysfunction (Kodani, 2023).
Fatty acid hydroxylase-associated neurodegeneration (FAHN) is a rare disease that exhibits brain modifications and motor dysfunctions in early childhood. The condition is caused by a homozygous or compound heterozygous mutation in fatty acid 2 hydroxylase (FA2H), whose encoded protein synthesizes 2-hydroxysphingolipids and 2-hydroxyglycosphingolipids and is therefore involved in sphingolipid metabolism. Drosophila is an excellent model for many neurodegenerative disorders; hence, this study has characterized and validated the first FAHN Drosophila model. The investigation of loss of dfa2h lines revealed behavioral abnormalities, including motor impairment and flying disability, in addition to a shortened lifespan. Furthermore, alterations in mitochondrial dynamics, and autophagy were identified. Analyses of patient-derived fibroblasts, and rescue experiments with human FA2H, indicated that these defects are evolutionarily conserved. This study thus presents a FAHN Drosophila model organism that provides new insights into the cellular mechanism of FAHN (Mandik, 2022).
Brain aging may accelerate after rodents reach middle age. However, the endogenous mediator that promotes this acceleration is unknown. It is predicted that the mediator may be expressed after an organism reaches middle age and dysregulates mitochondrial function. In the neurons of wild-type Drosophila (flies), it was observed that mitochondria were fragmented in aged flies, and this fragmentation was associated with mitochondrial calcium overload. In a previous study, it was found that mitochondrial fragmentation induced by calcium overload was reversed by the loss of Vimar, which forms a complex with Miro. Interestingly, Vimar expression was increased after the flies reached middle age. Overexpression of Vimar in neurons resulted in premature aging and mitochondrial calcium overload. In contrast, downregulation of Vimar in flies older than middle age promoted healthy aging. As the mouse homolog of Vimar, RAP1GDS1 expression was found to be increased after mice reached middle age; RAP1GDS1-transgenic and RAP1GDS1-knockdown mice displayed similar responses to flies with overexpressed and reduced Vimar expression, respectively. This research provides genetic evidence of a conserved endogenous mediator that promotes accelerated brain aging (Xiong, 2023).
Aberrant immune responses and chronic inflammation can impose significant health risks and promote premature aging. Pro-inflammatory responses are largely mediated via reactive oxygen species (ROS) and reduction-oxidation reactions. A pivotal role in maintaining cellular redox homeostasis and the proper control of redox-sensitive signaling belongs to a family of antioxidant and redox-regulating thiol-related peroxidases designated as peroxiredoxins (Prx). Recent studies in Drosophila have shown that Prxs play a critical role in aging and immunity. This study identified two important 'hubs', the endoplasmic reticulum (ER) and mitochondria, where extracellular and intracellular stress signals are transformed into pro-inflammatory responses that are modulated by the activity of the Prxs residing in these cellular organelles. This study found that mitochondrial Prx activity in the intestinal epithelium is required to prevent the development of intestinal barrier dysfunction, which can drive systemic inflammation and premature aging. Using a redox-negative mutant, it ws demonstrated that Prx acts in a redox-dependent manner in regulating the age-related immune response. The hyperactive immune response observed in flies under-expressing mitochondrial Prxs is due to a response to abiotic signals but not to changes in the bacterial content. This hyperactive response, but not reduced lifespan phenotype, can be rescued by the ER-localized Prx (Odnokoz, 2023).
In a global aging population, it is important to understand the factors affecting systemic aging and lifespan. Mitohormesis, an adaptive response caused by different insults affecting the mitochondrial network, triggers a response from the nuclear genome inducing several pathways that promote longevity and metabolic health. Understanding the role of mitochondrial function during the aging process could help biomarker identification and the development of novel strategies for healthy aging. This study interfered the muscle expression of the Drosophila genes Marf and Opa1, two genes that encode for proteins promoting mitochondrial fusion, orthologues of human MFN2 and OPA1. Silencing of Marf and Opa1 in muscle increases lifespan, improves locomotor capacities in the long term, and maintains muscular integrity. A metabolomic analysis revealed that muscle down-regulation of Marf and Opa1 promotes a non-autonomous systemic metabolome reorganization, mainly affecting metabolites involved in the energetic homeostasis: carbohydrates, lipids and aminoacids. Interestingly, the differences are consistently more evident in younger flies, implying that there may exist an anticipative adaptation mediating the protective changes at the older age. This study demonstrates that mild mitochondrial muscle disturbance plays an important role in Drosophila fitness and reveals metabolic connections between tissues. This study opens new avenues to explore the link of mitochondrial dynamics and inter-organ communication, as well as their relationship with muscle-related pathologies, or in which muscle aging is a risk factor for their appearance. These results suggest that early intervention in muscle may prevent sarcopenia and promote healthy aging (Tapia, 2021).
The Drosophila bang-sensitive mutant tko25t, manifesting a global deficiency in oxidative phosphorylation due to a mitochondrial protein synthesis defect, exhibits a pronounced delay in larval development. Previous work has identified a number of metabolic abnormalities in tko25t larvae, including elevated pyruvate and lactate, and found the larval gut to be a crucial tissue for the regulation of larval growth in the mutant. This study established that expression of wild-type tko in any of several other tissues of tko25t also partially alleviates developmental delay. The effects appeared to be additive, whilst knockdown of tko in a variety of specific tissues phenocopied tko25t, producing developmental delay and bang-sensitivity. These findings imply the existence of a systemic signal regulating growth in response to mitochondrial dysfunction. Drugs and RNAi-targeted on pyruvate metabolism interacted with tko25t in ways that implicated pyruvate or one of its metabolic derivatives in playing a central role in generating such a signal. RNA-seq revealed that dietary pyruvate-induced changes in transcript representation were mostly non-coherent with those produced by tko25t or high-sugar, consistent with the idea that growth regulation operates primarily at the translational and/or metabolic level (George, 2019).
Mitochondria provide energy for cellular function. This study examined daily changing patterns of mitochondrial function and metabolism in Drosophila in vivo in terms of their complex (I-IV) activity, ATP production, glycolysis, and whole fly respiration in the morning, afternoon and night. Complex activity and respiration showed significant and unexpected variation, peaking in the afternoon. However, ATP levels by contrast are >40% greater in the morning and lowest at night when glycolysis peaks. Complex activity modulation was at the protein level with no evidence for differential transcription over the day. Timing differences between increased ATP production and peaks of complex activity may result from more efficient ATP production early in the day leaving complex activity with spare capacity. Optical stimulation of mitochondria is only possible in the mornings when there is such spare capacity. These results provide first evidence of shifts in cellular energy capacity at the organism level. Understanding their translation may be significant to the chosen timing of energy demanding interventions to improve function and health (Weinrich, 2019).
Drosophila Prominin-like is a homolog of mammalian CD133, which is recognized as a biomarker for stem cells. This study found that Drosophila Prominin-like interacts with ND-20, a subunit of mitochondrial respiratory complex I. Prominin-like is a six-transmembrane glycoprotein which localizes on cellular membranes. Prominin-like localizes in the mitochondria. The knockdown of prominin-like in S2 cells resulted in transient mitochondrial dysfunctions as evidenced by reduced ATP production, elevated ROS generation and an accompanied reduction in mitochondrial proteins. Mitochondrial dysfunctions were detected in aged prominin-like mutant flies. The data indicates that Prominin-like acts to maintain mitochondrial function through its interaction with ND-20 which, itself, is active in the mitochondrial electron transport chain (Wang, 2019).
Phosphatidylserine (PS), synthesized in the endoplasmic reticulum (ER) by phosphatidylserine synthetase (PSS; CG4825), is transported to the plasma membrane (PM) and mitochondria through distinct routes. The in vivo functions of PS at different subcellular locations and the coordination between different PS transport routes are not fully understood. This paper reports that Drosophila PSS regulates cell growth, lipid storage and mitochondrial function. In pss RNAi, reduced PS depletes plasma membrane Akt, contributing to cell growth defects; the metabolic shift from phospholipid synthesis to neutral lipid synthesis results in ectopic lipid accumulation; and the reduction of mitochondrial PS impairs mitochondrial protein import and mitochondrial integrity. Importantly, reducing PS transport from the ER to PM by loss of PI4KIIIalpha partially rescues the mitochondrial defects of pss RNAi. Together, these results uncover a balance between different PS transport routes and reveal that PSS regulates cellular homeostasis through distinct metabolic mechanisms (Yang, 2019)
Evolutionary theory proposes that maternal inheritance of mitochondria will facilitate the accumulation of mitochondrial DNA (mtDNA) mutations that are harmful to males but benign or beneficial to females. It is predicted that the genetic variation which delineates distinct mtDNA haplotypes of a given species should confer larger phenotypic effects on males than females (reflecting mtDNA mutations that are male-harming, but female-benign), or sexually antagonistic effects (reflecting mutations that are male-harming, but female-benefitting). This study explored whether similar signatures of male-bias or sexual antagonism extend to a key physiological trait-metabolic rate. The effects of mitochondrial haplotypes on the amount of carbon dioxide produced by individual flies, controlling for mass and activity, was measured across 13 strains of D. melanogaster that differed only in their mtDNA haplotype. The effects of mtDNA haplotype on metabolic rate were larger in males than females. Furthermore, a negative intersexual correlation was observed across the haplotypes for metabolic rate. Finally, a male-specific negative correlation, across haplotypes, was uncovered between metabolic rate and longevity. These results are consistent with the hypothesis that maternal mitochondrial inheritance has led to the accumulation of a sex-specific genetic load within the mitochondrial genome, which affects metabolic rate and that may have consequences for the evolution of sex differences in life history (Nagarajan-Radha, 2020).
Oogenesis features an enormous increase in mitochondrial mass and mtDNA copy number, which are required to furnish mature eggs with an adequate supply of mitochondria and to curb the transmission of deleterious mtDNA variants. Quiescent in dividing germ cells, mtDNA replication initiates upon oocyte determination in the Drosophila ovary, which necessitates active mitochondrial respiration. However, the underlying mechanism for this dynamic regulation remains unclear. This study shows that an feedforward insulin-Myc loop promotes mitochondrial respiration and biogenesis by boosting the expression of electron transport chain subunits and of factors essential for mtDNA replication and expression, and for the import of mitochondrial proteins. Transient activation of JNK enhances the expression of the insulin receptor and initiates the insulin-Myc signaling loop. This signaling relay promotes mitochondrial biogenesis in the ovary, and thereby plays a role in limiting the transmission of deleterious mtDNA mutations. This study demonstrates cellular mechanisms that couple mitochondrial biogenesis and inheritance with oocyte development (Wang, 2019).
Mitochondria host a number of biosynthetic pathways and produce most of the cell's ATP through oxidative phosphorylation, which is carried out by the electron transport chain (ETC) complexes located on the mitochondrial inner membrane. While the majority of mitochondrial proteins are encoded on the nuclear genome, synthesized in the cytoplasm, and imported into the mitochondria, a subset of core ETC components are encoded on the mitochondrial genome (mtDNA) and synthesized inside the mitochondrial matrix. Thus, mitochondria biogenesis and ETC activity in particular, rely on the coordinated expression of both nuclear- and mtDNA-encoded mitochondrial genes. Mitochondria vary in number and activity to meet the different energy and metabolic demands of different tissues and developmental processes. Mitochondria are transmitted exclusively through the maternal lineage in most metazoans, which demands a complex regulation of mitochondrial biogenesis and ETC activity during oogenesis. Animal oocytes are hundreds of times larger than their progenitors. During this tremendous oocyte growth, mitochondria undergo prodigious biogenesis and increase mtDNA copy number over a thousand folds. The massive amount of mitochondria in the mature oocyte is necessary to power early embryonic development, as inadequate mitochondrial contents often lead to embryonic lethality. However, the mechanism by which the germline couples mitochondrial biogenesis to oocyte development remains elusive (Wang, 2019).
While furnishing mature oocytes with sufficient number of mitochondria, oogenesis also limits the transmission of harmful mtDNA mutations. The mitochondrial genome is prone to accumulating mutations because of its close vicinity to the highly mutagenic free radicals present in the mitochondrial matrix and of a lack of effective repair mechanisms. Yet, harmful mtDNA mutations are rare in populations, underscoring the presence of efficient mechanisms to limit their transmission through the female germline. It has been reported that mtDNA replication depends on active respiration in the Drosophila ovary. Healthy mitochondria with wild-type genomes propagate more vigorously than defective ones carrying harmful mutations, thereby curbing the transmission of deleterious mtDNA mutations to the next generation. Therefore, an active ETC appears to be a stress test for the functionality of mtDNA, and is essential for mtDNA selective inheritance. Nonetheless, how the activity of the ETC is regulated during oogenesis is not well understood (Wang, 2019).
Insulin signaling (IIS), an evolutionary conserved pathway that controls cell growth and proliferation, has also been shown to regulate ETC biogenesis and ATP production in human skeletal muscles. In the Drosophila ovary, IIS promotes the growth of follicles from the early to the middle stages of oogenesis. IIS activity decreases before the nurse cells dump their content into the oocyte. This decrease relieves the inhibition of GSK3, thereby shutting down mitochondrial respiration. However, oogenesis begins with germline stem cells (GSCs) that are thought not to rely on oxidative phosphorylation to ATP production. It is predicted there had to be developmental cues to activate mitochondrial respiration in the late germarium stage when mtDNA replication commences. IIS represents a logical candidate to modulate this metabolic transition in early oogenesis. Nonetheless, it remains to be explored how IIS is dynamically regulated during oogenesis and whether it is indeed involved in the aforementioned metabolic transition. Furthermore, little is known regarding how IIS modulates ETC activity and mtDNA biogenesis in general (Wang, 2019).
This study found that mitochondrial respiration is quiescent in GSCs and dividing cysts, but markedly upregulated in the late germarium, the same spatial-temporal pattern as mtDNA replication. A feedforward loop was found between IIS and Myc protein which orchestrates the transcriptional activation of respiration and mtDNA replication. Furthermore, transient JNK activity boosts insulin receptor (InR) transcription to enhance the IIS-Myc loop. This work uncovers how developmental programs couple mitochondrial biogenesis with cell growth and mitochondrial inheritance (Wang, 2019).
mtDNA replication in the Drosophila ovary relies on active respiration, suggesting that ETC activity and mtDNA replication might be subject to the same spatio-temporal regulation. This study has addressed this question and has further elucidated the developmental mechanisms regulating ETC activity and mtDNA biogenesis in the ovary. Utilizing the COX/SDH dual activity staining, it was revealed that ETC complexes are inactive in the germline stem cells (GSCs) and dividing cysts from germarium region 1 to 2A, but sharply activated in region 2B and active through stage-10 follicles. This spatial pattern mirrors that of mtDNA replication in the Drosophila ovary, supporting an essential role of mitochondrial respiration in mtDNA inheritance, both quantitively and qualitatively. It was also demonstrated that ETC activation is accompanied with an upregulation of the expression of ETC genes of both nuclear and mitochondrial origin. Interestingly, MDI, which drives the local translation of nuclear encoded mitochondrial proteins on the mitochondrial outer membrane and TFAM, which governs mtDNA replication and transcription, exhibit the same developmental pattern as mitochondrial respiration in the germarium. Collectively, these proteins would boost the biogenesis of ETC in region 2B of the germarium and in growing egg chambers. In an ovariole, different stages of developing germ cells reside in the same microenvironment and experience the same oxygen tension. Thus, the mitochondrial respiratory activity is likely to be determined by the abundance of ETC components, which itself is controlled by transcriptional activationx (Wang, 2019).
To understand how mitochondrial respiration is regulated, an RNAi screen was conducted for genes that boost COX/SDH activity in the ovary. The myc gene emerged as one of the strongest hits, and a hypomorphic allele, mycP0, largely abolished ETC activity and mtDNA replication in the germarium. Moreover, the spatial pattern of Myc protein mirrors mtDNA replication and ETC activity, further supporting its essential role in transcriptional activation of ETC biogenesis. RNA sequencing data demonstrate that Myc broadly stimulates gene expression in the Drosophila ovary, including many nuclear-encoded ETC genes and factors required for mtDNA replication and expression. These observations are consistent with previous studies in mammals showing that MYC can promote mitochondrial biogenesis by directly elevating the expression of nuclear-encoded mitochondrial genes. Among 198 annotated human mitochondrial genes that are up-regulated by Myc overexpression, 185 have homologs in the Drosophila genome. Of note, 44.9% (101 out of 225) of the fly homologs are down-regulated in mycP0 mutant ovaries, suggesting an evolutionarily conserved function of Myc in regulating mitochondrial biogenesis through gene expression. The finding that Myc induces ETC biogenesis and respiration is also in line with the studies in mammals demonstrating the multi-faceted roles of Myc in the regulation of mitochondria, including boosting mitochondrial biogenesis, stimulating oxidative metabolism , and regulating mitochondrial structure and dynamics (Wang, 2019).
Myc overexpression sometimes gives rise to different transcriptional output in different cell types. This observation reflects the fact that Myc-family proteins often associate with other cofactors and exert a broad and complex transcriptional role in a cell- or tissue-specific manner. This study also found that 130 transcription regulators, including Spargel Srl (fly homolog of human PGC-1) and CG32343 (fly homolog of GABPB2), were affected by the mycP0 mutation. PGC-1 proteins belong to an evolutionarily conserved family that integrates mitochondrial biogenesis and energy metabolism with a variety of cellular processes. In Drosophila, Srl regulates the expression of a subset of nuclear encoded mitochondrial genes. Mammalian GABPB2 is a regulatory subunit of the Nuclear Respiratory Factor complex 2 that regulates the expression of a small set of nuclear encoded mitochondrial proteins. Therefore, additional tiers of transcriptional regulations downstream of Myc are likely involved in boosting ETC biogenesis (Wang, 2019).
While myc transcription is uniform in the germarium, Myc protein is elevated at region 2B and remains high until the stage-10 egg chamber, indicating that Myc abundance is mainly regulated via post transcriptional mechanisms. IIS and JNK also emerged from the RNAi screen, and both were further confirmed to be required for triggering ETC biogenesis and mtDNA replication. IIS activity, marked by both p-AKT and p-GSK3 staining, displayed a pattern similar to that of Myc. Additionally, elevated IIS activity was required to establish a high level of Myc and to activate ETC in the late germarium stage. GSK3 directly phosphorylates Myc and promotes its ubiquitination and degradation in both mammalian and fly cultured cells. Thus, IIS likely stabilizes Myc protein by inhibiting GSK activity. This result is also in line with a previous study showing that decreased IIS activity relieves the inhibition on GSK3, which leads to mitochondrial quiescence at later stages of oogenesis. Importantly, this work uncovers Myc as the downstream effector of IIS in the regulation of respiration and mtDNA biogenesis in the ovary (Wang, 2019).
It was noticed that InR transcription was down-regulated in the myc mutant ovary, suggesting a positive feedback regulation between IIS and Myc. This regulatory loop maintains high levels of both Myc protein and IIS activity in the mid-stage follicles, where massive mitochondrial biogenesis and massive cell growth take place. However, it does not explain how this loop is activated in the first place at the late germarium stages. It was found that JNK was transiently activated in germ cells in the germarium region 2B, but decreased in budding egg chambers and sharply diminished thereafter. High level and sustained JNK activity often lead to apoptosis. However, cell death is rarely observed in the germaria of flies cultured under normal conditions. Thus, JNK activation in the late germarium must be triggered by cellular processes unrelated to apoptosis. Transiently elevated JNK activity was sufficient to increase InR mRNA level, which in-turn boosted IIS activity and stabilized Myc protein. Currently, the link between JNK and IIS is not well-understood. In the metastatic Drosophila epithelium, cell survival and proliferation entail upregulation of InR expression by JNK through wingless signaling. However, no genes in the wingless signaling pathway emerged from the RNAi screen in germ cells. The molecular mechanisms that links JNK activation to InR expression in ovary remain to be explored (Wang, 2019).
The JNK-dependent transcriptional program can be activated by various cellular stresses and cell-cell signaling events. In region 2B of the germarium, the follicle cells extend and migrate laterally across the germarium to wrap around the 16 cells cyst. Thus, JNK activation in germ cells may reflect paracrine signaling from the follicle cells, for instance via TNF-α. Alternatively, the process of follicle cells enveloping and compressing the 16-cell cyst may generate mechanical stress that subsequently activates JNK. Regardless, this work uncovers a novel function of JNK in energy metabolism and mitochondrial biogenesis besides its well-established roles in controlling cell apoptosis, growth, and proliferation (Wang, 2019).
Studies in a variety of animal models have shown that reproductive aging in females is tightly associated with decreased IIS activity. Interestingly, oocytes of aged females often have higher incidence of mtDNA lesions and lower mtDNA copy number. Thus, developmental control of mitochondrial biogenesis and mtDNA replication via IIS may be a conserved mechanism in metazoans. Previous studies demonstrated that prodigious mitochondrial biogenesis during oogenesis underlies the selective inheritance of functional mtDNA by allowing proliferation competition between healthy mitochondria and mitochondria carrying deleterious mtDNA mutations. This study has shown that the JNK/IIS/Myc signaling relay governs mitochondrial biogenesis in the ovary, and thereby influences mitochondrial inheritance both quantitively and quantitively. These studies could provide a molecular framework to further understand the control of mitochondrial biogenesis and mtDNA inheritance in animals (Wang, 2019).
Mitochondria generate ATP and building blocks for cell growth and regeneration, using pyruvate as the main substrate. This study introduced PyronicSF, a user-friendly GFP-based sensor of improved dynamic range that enables real-time subcellular quantitation of mitochondrial pyruvate transport, concentration and flux. Cultured mouse astrocytes were shown to maintain mitochondrial pyruvate in the low micromolar range, below cytosolic pyruvate, which means that the mitochondrial pyruvate carrier MPC is poised to exert ultrasensitive control on the balance between respiration and anaplerosis/gluconeogenesis. The functionality of the sensor in living tissue is demonstrated in the brain of Drosophila melanogaster larvae. Mitochondrial subpopulations are known to coexist within a given cell, which differ in their morphology, mobility, membrane potential, and vicinity to other organelles. The present tool can be used to investigate how mitochondrial diversity relates to metabolism, to study the role of MPC in disease, and to screen for small-molecule MPC modulators (Arce-Molina, 2020).
The endosymbiotic theory proposes that eukaryotes evolved from the symbiotic relationship between anaerobic (host) and aerobic prokaryotes. Through iterative genetic transfers, the mitochondrial and nuclear genomes coevolved, establishing the mitochondria as the hub of oxidative metabolism. To study this coevolution, this study disrupt mitochondrial-nuclear epistatic interactions by using strains that have mitochondrial DNA (mtDNA) and nuclear DNA (nDNA) from evolutionarily divergent species. A multifaceted approach was undertaken generating introgressed Drosophila strains containing D. simulans mtDNA and D. melanogaster nDNA with Sirtuin 4 (Sirt4)-knockouts. Sirt4 is a nuclear-encoded enzyme that functions, exclusively within the mitochondria, as a master regulator of oxidative metabolism. Flies were exposed to the drug rapamycin in order to eliminate TOR signaling, thereby compromising the cytoplasmic crosstalk between the mitochondria and nucleus. The results indicate that D. simulans and D. melanogaster mtDNA haplotypes display opposite Sirt4-mediated phenotypes in the regulation of whole-fly oxygen consumption. Moreover, the data reflect that the deletion of Sirt4 rescued the metabolic response to rapamycin among the introgressed strains. It is proposed that Sirt4 is a suitable candidate for studying the properties of mitochondrial-nuclear epistasis in modulating mitochondrial metabolism (Sejour, 2020).
Reactive Oxygen Species (ROS) are essential cellular messengers required for cellular homeostasis and regulate the lifespan of several animal species. The main site of ROS production is the mitochondrion, and within it, respiratory complex I (CI) is the main ROS generator. ROS produced by CI trigger several physiological responses that are essential for the survival of neurons, cardiomyocytes and macrophages. This study show that CI produces ROS when electrons flow in either the forward (Forward Electron Transport, FET) or reverse direction (Reverse Electron Transport, RET). ROS production via RET (ROS-RET) is activated under thermal stress conditions, and interruption of ROS-RET production, through ectopic expression of the alternative oxidase AOX, attenuates the activation of pro-survival pathways in response to stress. Accordingly, this study found that both suppressing ROS-RET signalling or decreasing levels of mitochondrial H2O2 by overexpressing mitochondrial catalase (mtCAT), reduces survival dramatically in flies under stress. These results uncover a specific ROS signalling pathway where hydrogen peroxide (H2O2) generated by CI via RET is required to activate adaptive mechanisms, maximising survival under stress conditions (Scialo, 2020).
Mitochondrial dysfunctions belong amongst the most common metabolic diseases but the signalling networks that lead to the manifestation of a disease phenotype are often not well understood. This study identified the subunits of respiratory complex I, III and IV as mediators of major signalling changes during Drosophila wing disc development. Their downregulation in larval wing disc leads to robust stimulation of TOR activity, which in turn orchestrates a complex downstream signalling network. Specifically, after downregulation of the complex I subunit ND-49 (mammalian NDUFS2), TOR activates JNK to induce cell death and ROS production essential for the stimulation of compensatory apoptosis-induced proliferation within the tissue. Additionally, TOR upregulates Notch and JAK/STAT signalling and it directs glycolytic switch of the target tissue. These results highlight the central role of TOR signalling in mediating the complex response to mitochondrial respiratory dysfunction and they provide a rationale why the disease symptoms associated with respiratory dysfunctions are often alleviated by mTOR inhibitors (Perez-Gomez, 2020).
Mitochondria play an essential function in cellular energetic and NADH metabolism. Five protein complexes (complex I-V) of the electron transport chain (ETC) have distinct functions in the oxidation of NADH and/or FADH2, maintenance of the inner mitochondrial membrane potential and production of ATP via oxidative phosphorylation. Moreover, they serve as signalling hubs for specific cellular events including ROS-mediated signalling, apoptosis and Ca2+ signalling. Mutations in mitochondrial enzymes are the most frequent metabolic mutations present in human (Perez-Gomez, 2020).
Complex I of the ETC is the node point in the mitochondrial NADH metabolism as it mediates electron transfer from NADH to the other respiratory complexes. Therefore, complex I inhibitors have been exploited as therapeutic targets in cancer treatment, although the mechanism of action is often unclear. On the other hand, complex I inhibition can lead to increased proliferation, depending on the cell type and complex I inhibitor used. Despite the fact that mitochondrial electron transport chain disorders are one of the most common human genetic diseases, the mechanisms behind the dichotomy in the functional outputs after complex I inhibition are not well understood (Perez-Gomez, 2020).
The mTOR pathway (TOR in Drosophila) is the key integrator of cellular metabolic inputs that connects cell growth with environmental signals, including nutrient and growth factors availability. It promotes cell growth by stimulation of cellular translation, anabolic metabolism and by inhibiting autophagy. At the same time, mTOR activation can lead to apoptosis in certain contexts. The upregulation of mTOR is observed during epithelial wound healing, during aging as well as in many types of cancers. Although strong inhibition of mitochondrial respiration can cause a metabolic catastrophe and cell death connected with mTOR inhibition through the activation of AMPK, increasing evidence suggest that many types of respiratory dysfunctions are connected with increase in mTOR activity and mTOR inhibition leads to alleviation of the phenotype (Perez-Gomez, 2020).
This study found that downregulation of mitochondrial respiratory complex I, III or IV stimulates TOR activity that directs major downstream signalling events, including Notch activation, metabolic changes and apoptosis-driven proliferation. As TOR overactivation balances between stimulation of apoptosis and proliferation, presented in this study suggests a possible mechanism for the observations when complex I inhibition promotes either cell death or proliferation in different contexts. The signalling network that was identified also suggests a possible explanation why the disease symptoms associated with respiratory dysfunctions are often alleviated by mTOR inhibitors (Perez-Gomez, 2020).
Despite the fact that mutations in mitochondrial enzymes are the most frequent metabolic mutations present in human, manifested in a whole range of clinical disorders, the actual signalling networks that are triggered by malfunctioning mitochondria to develop clinical symptoms are still not well understood. The results argue that TOR pathway is the key signalling effector triggered after downregulation of complex I, III or IV in the Drosophila wing disc. TOR activity in turn activates JNK, Notch and JAK/STAT signalling, boosts glycolysis and promotes compensatory apoptosis-induced proliferation to produce profound effects on tissue size and patterning (Perez-Gomez, 2020).
By placing TOR at the top of the signalling network triggered by complex I dysfunction, this study provides a rationale for numerous observations where TOR inhibition alleviated the disease symptoms associated with mitochondrial dysfunction. For example, the maternally inherited Leigh syndrome (MILS), caused by mutation in complex I subunit Ndufs4, is associated with enhanced mTOR activity in neurons and the disease symptoms can be alleviated using chemical mTOR inhibitors. Upregulation of mTORC1 has also been described as a key component of the mitochondrial integrated stress response during mitochondrial myopathy. Alongside this line, the low survival rate of flies with mutation in the ND2 subunit of complex I can also be rescued by chemical TOR inhibition in Drosophila. Moreover, an aggressive phenotype of breast cancer that is associated with complex I mutations can be reversed via restoration of complex I function that is associated with decreased mTOR activity. Therefore, the data showing the role of TOR at the very apex of the signalling hierarchy after complex I dysfunction makes it interesting to test if similar regulatory mechanism underpins other types of mitochondrial dysfunction (Perez-Gomez, 2020).
Respiratory inhibitors are used to surpress various types of cancers although respiratory dysfunction can also promote cancer progression. Based on the current data it can be hypothesized that the types of complex I dysfunctions that stimulate cancer progression would correlate with overstimulation of mTOR activity that initiates downstream signalling events promoting apoptosis but also apoptosis-induced proliferation. This may seem contradictory as complex I inhibition usually leads to decrease in ATP:ADP ratio that in turn activates AMPK, a known suppressor of mTOR activity. However, the evidence that complex I inhibition would actually lead to downregulation of mTOR activity is surprisingly scarce and concerns mainly complex I inhibitors like biguanides (metformin, fenformin) or fenofibrate. On the contrary, there is ample of observations where mTOR is increased during mitochondrial dysfunction, supporting the results. In fact, mTOR activity can be stimulated even in the presence of active AMPK, as in the case of the Leigh syndrome caused by mutation in complex I subunit Ndufs4. By balancing between stimulation of apoptosis and proliferation, the mTOR driven signalling network identified in this study may suggest a possible mechanism for the contradictory observations where complex I inhibition was reported to promote cell death but also support proliferation depending on context (Perez-Gomez, 2020).
One remaining question is how TOR is upregulated by mitochondria. One possibility may be an activation of TOR via mitochondrial Akt signalling and TOR complexes located in mitochondria-associated endoplasmic reticulum. However, since TOR operates at the top of the signalling hierarchy after complex I downregulation, it can also be speculated that its activity could be sensitive to the primary metabolic misbalance caused by disruption of mitochondrial metabolism. Indeed, the decrease in the NAD:NADH ratio and the associated slowdown of the TCA cycle that are associated with downregulation of respiration are likely to influence the activity of protein metabolic sensors such as sirtuin deacetylases or 2-KG dependent demethylases that may in turn regulate mTOR activity, either directly or indirectly, as suggested in some other contexts (Perez-Gomez, 2020).
Through non-apoptotic roles of caspases, dying cells can release diffusible mitogens and thus signal to their neighbours and instruct them to proliferate -- a process known as apoptosis-induced proliferation (AIP). Several modes of AIP have been described in various species and tissues characterised by the use of either initiator or effector caspase to drive the signalling mechanism that in turn promotes proliferation. In Drosophila wing or eye discs, the most studied AIP models are based on targeted expression of the pro-apoptotic genes hid or rpr where the proliferation is dependent on the initiator caspases that activate ROS production and JNK activity. In this model, proliferation is also dependent on caspases, ROS production and JNK activation, however it is unique in the way it is triggered and in the way the signalling components are interconnected: (1) it is initiated by downregulation of mitochondrial respiratory complex I and therefore it has a metabolic origin (2) it is orchestrated by consequent activation of the TOR pathway (3) it is dependent on effector caspases and (4) JNK activation is upstream of cell death, not activated by the non-apoptotic roles of caspases. Examples of AIP dependent on effector caspases have been described in Drosophila postmitotic cells in the eye disc and in mammalian cells after irradiation induced cell damage but the signalling components involved and their regulatory relationship also differ from the model used in this study (Perez-Gomez, 2020).
It is important to stress that the high levels of ROS species are not produced in every cell where ND-49-RNAi is induced. Although low levels of ROS may appear as a primary consequence of ND-49 downregulation, the strong ROS signal observed in certain areas of the wing disc occurs downstream of cell death, as blocking apoptosis alongside ND-49-RNAi also eliminates the ROS signal. This is in agreement with ROS generation in other modes of AIP. Although ROS production was described with certain complex I inhibitors it does not happen when other inhibitors are used. Assembly of complex I into supercomplexes with other ETC proteins determines if ROS will be produced or not. In the current model it is obvious that the majority of ROS observed does not originate from the dysfunctional complex I but they result from apoptosis, as blocking apoptosis prevents also ROS formation (Perez-Gomez, 2020).
Taken together, the results highlight the central role of TOR pathway activation during mitochondrial dysfunction. As TOR overactivation gives identical phenotype to complex I downregulation, future studies should investigate if the results may be relevant outside of the mitochondria field, in some of the other contexts involving TOR overactivation, such as many types of cancer, wound healing or aging, with potentially important clinical implications (Perez-Gomez, 2020).
In insect, pyruvate is generally the predominant oxidative substrate for mitochondria. This metabolite is transported inside mitochondria via the mitochondrial pyruvate carrier (MPC), but whether and how this transporter controls mitochondrial oxidative capacities in insects is still relatively unknown. This study characterized the importance of pyruvate transport as a metabolic control point for mitochondrial substrate oxidation in two genotypes of an insect model, Drosophila melanogaster, differently expressing MPC1, an essential protein for the MPC function. The kinetics were evaluated of pyruvate oxidation, mitochondrial oxygen consumption, metabolic profile, activities of metabolic enzymes, and climbing abilities of wild-type (WT) flies and flies harboring a deficiency in MPC1 (MPC1(def)). It was hypothesized that MPC1 deficiency would cause a metabolic reprogramming that would favor the oxidation of alternative substrates. The results show that the MPC1(def) flies display significantly reduced climbing capacity, pyruvate-induced oxygen consumption, and enzymatic activities of pyruvate kinase, alanine aminotransferase, and citrate synthase. Moreover, increased proline oxidation capacity was detected in MPC1(def) flies, which was associated with generally lower levels of several metabolites, and particularly those involved in amino acid catabolism such as ornithine, citrulline, and arginosuccinate. This study therefore reveals the flexibility of mitochondrial substrate oxidation allowing Drosophila to maintain cellular homeostasis (Simard, 2020).
The mitochondrial electron transport chain (ETC) enables essential metabolic reactions; nonetheless, the cellular responses to defects in mitochondria and the modulation of signaling pathway outputs are not understood. This study shows that Notch signaling and ETC attenuation via knockdown of COX7a induces massive over-proliferation. The tumor-like growth is caused by a transcriptional response through the eIF2α-kinase PERK and ATF4, which activates the expression of metabolic enzymes, nutrient transporters, and mitochondrial chaperones. This stress adaptation is found to be beneficial for progenitor cell fitness, as it renders cells sensitive to proliferation induced by the Notch signaling pathway. Intriguingly, over-proliferation is not caused by transcriptional cooperation of Notch and ATF4, but it is mediated in part by pH changes resulting from the Warburg metabolism induced by ETC attenuation. These results suggest that ETC function is monitored by the PERK-ATF4 pathway, which can be hijacked by growth-promoting signaling pathways, leading to oncogenic pathway activity (Sorge, 2020).
Controlling cell proliferation is one of the major challenges of multicellular life, both during phases of growth in developing organisms and phases of homeostatic cell replenishment essential in adult animals. Lack of appropriate control can lead to severe disorders, including cancer, at any stage of life. While over-proliferation of transformed, cancerous cells is usually caused by inactivation of tumor suppressors and/or activation of oncogenes, it has long been noted that tumors exhibit altered cellular characteristics such as a glycolytic metabolism. While this metabolic switch has been shown to be caused by oncogene signaling, cellular metabolism is also controlled at multiple levels under normal physiological (non-transformed) conditions, including at the transcriptional level through diverse stress-response pathways. One of these is the activating transcription factor 4 (ATF4), which is known to activate a transcriptional (integrated) stress response (ISR) under various stress conditions that trigger phosphorylation of eIF2α. The ATF4 transcriptional program consists of a diverse set of genes with cytoprotective function, but chronic activation induces apoptosis indirectly through transcription of the mammalian ATF4 target CHOP. Yet, ATF4 activation has been detected in several human tumors, especially in hypoxic or nutrient-deprived regions, where ATF4 has been attributed with pro-survival and pro-proliferative effects. Interestingly, a recent study showed that melanoma cells respond to inhibition of their glycolytic metabolism by activating an ATF4 response, whose metabolic reconfiguration allows these cells to continue oncogenic growth, together arguing that ATF4 can provide cancer cells with a metabolic flexibility that allows them to tolerate hypoxic and nutritional stress or cancer therapy aimed at metabolism. Among the many conditions activating ATF4, recent work with cultured cells showed that inhibition of mitochondrial function is linked to ATF4 translation and activity. However, from these and other studies, both the mechanistic basis and the in vivo implications of this response remain to be elucidated (Sorge, 2020).
This study shows that in the fruit fly, Drosophila melanogaster, genetic perturbation of the electron transport chain (ETC), which induces a Warburg-like metabolism, activates a transcriptional stress response mediated through the eIF2α-kinase PERK and ATF4 in eye progenitor cells of Drosophila larvae. Importantly, this in vivo stress response is activated under ETC knockdown conditions, in the absence of obvious mitochondrial dysfunction. Interestingly, these results show that the ATF4 transcriptional response, which by itself causes reduced fitness of progenitor cells, is hijacked by growth-promoting pathways like Notch or Ras, leading to increased cellular fitness and enhanced proliferation. The data furthermore suggest that the pH changes associated with ETC impairment resulting in a switch of the metabolism to aerobic glycolysis play an important role in progenitor over-proliferation. In sum, this study shows that ATF4-mediated transcriptional adaptation provides a cell-autonomous response to ETC defects, altering cellular behavior through metabolic adaptation (Sorge, 2020).
Genetically induced disturbance of ETC complex assembly resultrd in a metabolic shift typical for mitochondrial impairment and activated an ATF4-dependent stress response. The in vivo transcriptional adaptation presented in this study confirmed the regulation of LDH and glycolytic enzymes, as shown in Drosophila cultured cells, and further includes several targets shown to be ATF4 target genes in mammalian models. The results showed that the eIF2α-kinase PERK, so far only described for its role in mediating one branch of the unfolded protein response of the endoplasmic reticulum (UPRER), is the upstream kinase phosphorylating eIF2α, thereby inducing ATF4 translation in response to mitochondrial ETC disturbance. Mitochondrial ETC disturbance specifically activated PERK, while other branches of the UPRER were non-responsive. PERK activation upon mitochondrial defects was recently observed in Drosophila models of Parkinson's disease and was explained by the authors by its preferential localization to mitochondria-associated ER membranes, which might make PERK more susceptible to a local stress signal. ROS (reactive oxygen species) released by mitochondria have been suggested to mediate mitochondrial retrograde signaling. While this study observed an attenuation of Delta overexpression (DlOE), COX7RNAi-induced over-proliferation upon overexpression of either cytoplasmic catalase or GPx (but not mitochondrial catalase), this study failed to detect increased ROS levels in the larval eye disc. A possible scenario to explain these observations is that ROS are generated locally in the cytoplasm or ER in response to ETC disturbance, thereby triggering PERK activation. Importantly, Drosophila PERK isoform B contains a potential mitochondrial signal peptide, which is not found in mammalian PERK isoforms. Although no evidence for this has been found, Drosophila PERK could reside in the mitochondrial membrane and sense the folding status of mitochondrial complexes. This hypothesis could explain the evolutionary difference between mitochondrial defects and ATF4 induction, as this appears to require GCN2 but not PERK in mammals or to be triggered independently of a single eIF2α-kinase. In addition to canonical ATF4 target genes, ATF4-dependent upregulation of mitochondrial chaperones, a response classically referred to as the mitochondrial UPR (UPRmt) was observed. In C. elegans, mitochondrial chaperone induction upon stress is mediated by ATF4-like transcription factor Atfs-1, while the mammalian UPRmt has been shown to be regulated by another evolutionary-related transcription factor, ATF5. The current data now showed that Drosophila ATF4 is required cell autonomously for the induction of mitochondrial chaperones upon ETC subunit knockdown, implying that Drosophila might represent the evolutionary ancestral ISR-UPRmt regulation through a single ATF4-like transcription factor (Sorge, 2020).
The cooperation between ATF4 target genes and the Notch or Ras pathways in Drosophila imaginal progenitors raised the intriguing possibility that these or other oncogenic pathways could benefit from ATF4 activity in human cancers. Over the last decades, it had been demonstrated that human cancer cells are exposed to several stresses, including hypoxia, ROS, or limitations in nutrient availability. In order to survive these conditions and maintain their growth capacity, tumor cells activate responses like the HIF1α transcription factor axis. Though less well studied, an involvement of ATF4 in cancer has been suggested mostly through work with cultured cells. This study analyzed gene expression in human cancer samples of The Cancer Genome Atlas (TCGA) datasets using Cancer-RNaseq-Nexus and the human protein pathology atlas and found that many of the well-characterized direct ATF4 targets are upregulated in a variety of cancer types. Most strikingly, transcriptomes of kidney renal clear cell carcinoma showed progressive induction of ATF4 and many of its direct targets (EIF4EBP1, ASNS, TRIB3, and VEGFA) on the transcriptional level, which strongly correlated with a poor prognosis in this type of cancer. These data suggest that the ATF4-mediated ISR is used by cancer cells to adapt their metabolic repertoire, thereby sustaining fast growth under increasingly unfavorable conditions (Sorge, 2020).
A novel finding presented in this study was the discovery that ATF4-mediated transcriptional adaptation due to ETC impairment allowed eye progenitors to increase their proliferation in response to signals from the Notch and Ras pathways. The primary questions arising from this genetic interaction is how these signaling pathways can overcome the apparent cellular stress and reduction in proliferation and induce the opposite effect, a massively increased rate of proliferation. Several lines of evidence suggest that over-proliferation in DlOE, COX7RNAi eye imaginal discs is controlled by pH changes induced by LDH that modify the activity of Notch downstream effectors. First, in COX7a-depleted cells, the metabolism is switched to aerobic glycolysis, leading to an increased production of lactate due to the activity of LDH. And, consistent with an accumulation of this metabolic acid, this study found the intracellular pH to be reduced in COX7RNAi cells. Second, DlOE, COX7RNAi-mediated over-proliferation was rescued by ATF4 knockdown and, to a lesser extent, pH buffering, showing that intracellular pH changes (downstream of ATF4 and LDH) play an important role in proliferation control. Third, LDH phenocopies COX7RNAi, indicating that most of the cooperative effects of Dl overexpression and COX7a knockdown are mediated by the ATF4 target LDH. In the same line, expression of LDH as one of the many ATF4 targets was sufficient to drive Dl-expressing cells into over-proliferation, strongly suggesting that the processes downstream of LDH-in particular, the changes in intracellular pH-lead to a modification of the Notch pathway. Finally, a cooperation of Notch and the COX7a nuclear effector ATF4 on the transcriptional level was not observed, showing that the Notch pathway is not hyper-activated, but arguing that over-proliferation is due to changes in the activity of Notch downstream effectors. The next obvious question is how changes in intracellular pH can modify the activity of signaling pathways. It is known that the intracellular pH can control the protonation of specific histidine residues in proteins acting as pH sensors, leading to changes in protein properties. Importantly, it has been shown recently in chicken embryos that intracellular pH changes induced by a Warburg-like metabolism control the acetylation of the Wnt effector &betsa;-catenin, thereby mediating Wnt signaling activation. Thus, this study envisions that pH changes induced by LDH expression lead to a (non-enzymatic) modification of Notch effectors, thereby increasing fitness and proliferation rates of eye progenitor cells (Sorge, 2020).
This is a very attractive model; however, one result was puzzling. Although LDH is sufficient to induce over-proliferation when combined with the Notch pathway, no rescue (but an increase in the severity) of the DlOE, COX7RNAi phenotype was observed when LDH was selectively depleted. This obvious discrepancy can be explained by different hypotheses. One of them is based on a recent study showing that LDHA inhibition in melanoma cell lines also failed to impact cell proliferation, survival, or tumor growth. In this context, LDHA inhibition engaged the GCN2-ATF4 signaling axis to initiate an expansive pro-survival response, including the upregulation of the glutamine transporter SLC1A5 and glutamine uptake, as well as mTORC1 activation. Another hypothesis is based on the finding that a major driver of over-proliferation is the intracellular pH. Since LDH catalyzes the conversion of pyruvate to lactate (and back), reducing LDH levels will affect the ratio of lactate to pyruvate, leading to an increase of the even stronger metabolic acid pyruvate. It has been shown that pyruvate as lactate induces a concentration-dependent intracellular acidification. Thus, it could be envisioned that an enhancement of proliferation rates beyond those observed in DlOE, COX7RNAi cells is a consequence of pyruvate accumulation in the absence of LDH, which enhances the decrease in the intracellular pH, resulting in the increase in proliferation rates (Sorge, 2020).
Excess dietary carbohydrates are linked to dysregulation of metabolic pathways converging to mitochondria and metabolic inflexibility. This study determined the role of the mitochondrial pyruvate carrier (MPC) in the occurrence of this metabolic inflexibility in wild-type (WT) and MPC1-deficient (MPC1(def)) flies that were exposed to diets with different sucrose concentrations for 15-25 days (Standard Diet: SD, Medium-Sucrose Diet: MSD, and High-Sucrose Diet: HSD). The results showed that MPC1(def) flies had lower mitochondrial respiration rates than WT flies on the SD and MSD. However, when exposed to the HSD, WT flies displayed decreased mitochondrial respiration rates compared to MPC1(def) flies. WT flies exposed to the HSD also displayed increased proline contribution and slightly decreased MPC1 expression. Surprisingly, when fed the MSD and the HSD, few metabolites were altered in WT flies whereas MPC1(def) flies display significant accumulation of glycogen, glucose, fructose, lactate, and glycerol. Overall, this suggests that metabolic inflexibility starts to occur in WT flies after 15-25 days of exposure to the HSD whereas the MPC1(def) flies display metabolic inflexibility independently of the diet provided. This study thus highlights the involvement of MPC as an essential protein in Drosophila to maintain proper metabolic homeostasis during changes in dietary resources (Simard, 2020).
The m-AAA proteases play a critical role in the proteostasis of inner mitochondrial membrane proteins, and mutations in the genes encoding these proteases cause severe incurable neurological diseases. To further explore the biological role of the m-AAA proteases and the pathological consequences of their deficiency, a genetic approach was used in the fruit fly Drosophila melanogaster to inactivate the ATPase family gene 3-like 2 (AFG3L2) gene, which encodes a critical component of the m-AAA proteases. Null alleles of Drosophila AFG3L2 die early in development, but partial inactivation of AFG3L2 using RNAi allowed survival to the late pupal and adult stages of development. Flies with partial inactivation of AFG3L2 exhibited behavioral defects, neurodegeneration, accumulation of unfolded mitochondrial proteins, and diminished respiratory chain (RC) activity. Further work revealed that the reduced RC activity was primarily a consequence of severely diminished mitochondrial transcription and translation. These defects were accompanied by activation of the mitochondrial unfolded protein response (mito-UPR) and autophagy. Overexpression of mito-UPR components partially rescued the AFG3L2-deficient phenotypes, indicating that protein aggregation partly accounts for the defects of AFG3L2-deficient animals. This work suggests that strategies designed to activate mitochondrial stress pathways and mitochondrial gene expression could be therapeutic in the diseases caused by mutations in AFG3L2 (Pareek, 2020).
Metabolic inflexibility is a condition that occurs following a nutritional stress which causes blunted fuel switching at the mitochondrial level in response to hormonal and cellular signalling. Linked to obesity and obesity related disorders, chronic exposure to a high fat diet (HFD) in animal models has been extensively used to induce metabolic inflexibility and investigate the development of various metabolic diseases. However, many questions concerning the systemic and mitochondrial responses to metabolic inflexibility remain. This study investigated the global and mitochondrial variations following a 10-day exposure to a HFD in adult Drosophila melanogaster. The results show that following 10-day exposure to the HFD, mitochondrial respiration rates measured in isolated mitochondria at the level of complex I were decreased. This was associated with increased contributions of non-classical providers of electrons to the electron transport system (ETS) such as the proline dehydrogenase (ProDH) and the mitochondrial glycerol-3-phosphate dehydrogenase (mtG3PDH) alleviating complex I dysfunctions, as well as with increased ROS production per molecule of oxygen consumed. These results also show an accumulation of metabolites from multiple different metabolic pathways in whole adult Drosophila and a drastic shift in the lipid profile which translated into decreased proportion of saturated and monounsaturated fatty acids combined with an increased proportion of polyunsaturated fatty acids. Thus, these results demonstrate the various responses to the HFD treatment in adult Drosophila melanogaster that are hallmarks of the development of metabolic inflexibility and reinforce this organism as a suitable model for the study of metabolic disorders (Cormier, 2021)
Increased blue light exposure has become a matter of concern as it has a range of detrimental effects, but the mechanisms remain unclear. Mitochondria absorb short wavelength light but have a specific absorbance at 420nm at the lower end of the human visual range. This 420nm absorption is probably due to the presence of porphyrin. This study examined the impact of 420nm exposure on Drosophila melanogaster mitochondria and its impact on fly mobility. Daily 15 mins exposures for a week significantly reduced mitochondrial complex activities and increased mitochondrial inner membrane permeability, which is a key metric of mitochondrial health. Adenosine triphosphate (ATP) levels were not significantly reduced and mobility was unchanged. There are multiple options for energy/time exposure combinations, but single 420nm exposure of 3h was applied to increase the probability of an effect on ATP and mobility, and both were significantly reduced. ATP and mitochondrial membrane permeability recovered and over corrected at 72h post exposure. However, despite this, normal mobility did not return. Hence, the effect of short wavelengths on mitochondrial function is to reduce complex activity and increasing membrane permeability, but light exposure to reduce ATP and to translate into reduced mobility needs to be sustained (Kam, 2021).
Ageing is a major risk factor for many of the most prevalent diseases, including neurodegenerative diseases, cancer, and heart disease. As the global population continues to age, behavioural interventions that can promote healthy ageing will improve quality of life and relieve the socioeconomic burden that comes with an aged society. Exercise is recognised as an effective intervention against many diseases of ageing, but we do not know the stage in an individual's lifetime at which exercise is most effective at promoting healthy ageing, and whether or not it has a direct effect on lifespan. This study exercised w(1118) Drosophila melanogaster, investigating the effects of sex and group size at different stages of their lifetime, and recorded their lifespan. Climbing scores at 30 days were measured to record differences in fitness in response to exercise. The mitochondrial proteome was assessed of w(1118) Drosophila that had been exercised for one week, alongside mitochondrial respiration measured using high-resolution respirometry, to determine changes in mitochondrial physiology in response to exercise. This study found that age-targeted exercise interventions improved the lifespan of both male and female Drosophila, and grouped males exercised in late life had improved climbing scores when compared with those exercised throughout their entire lifespan. The proteins of the electron transport chain were significantly upregulated in expression after one week of exercise, and complex-II-linked respiration was significantly increased in exercised Drosophila. Taken together, our findings provide a basis to test specific proteins, and complex II of the respiratory chain, as important effectors of exercise-induced healthy ageing (Ebanks, 2021).
Cellular metabolism must adapt to changing NADPH glyoxylate reductase demands to enable homeostasis. During immune responses or cancer metastasis, cells leading migration into challenging environments require an energy boost, but what controls this capacity is unclear. A previously uncharacterized nuclear protein, Atossa (encoded by CG9005) was studied that supports macrophage invasion into the germband of Drosophila by controlling cellular metabolism. First, nuclear Atossa increases mRNA levels of Porthos, a DEAD-box protein, and of two metabolic enzymes, lysine-α-ketoglutarate reductase (LKR/SDH) and NADPH glyoxylate reductase (GR/HPR), thus enhancing mitochondrial bioenergetics. Then Porthos supports ribosome assembly and thereby raises the translational efficiency of a subset of mRNAs, including those affecting mitochondrial functions, the electron transport chain, and metabolism. Mitochondrial respiration measurements, metabolomics, and live imaging indicate that Atossa and Porthos power up OxPhos and energy production to promote the forging of a path into tissues by leading macrophages. Since many crucial physiological responses require increases in mitochondrial energy output, this previously undescribed genetic program may modulate a wide range of cellular behaviors (Emtenani, 2022).
Uncoupling proteins (UCPs) form a distinct subfamily of the mitochondrial carrier family (MCF) SLC25. Four UCPs, DmUCP4A-C and DmUCP5, have been identified in Drosophila melanogaster on the basis of their sequence homology with mammalian UCP4 and UCP5. In a Parkinson's disease model, DmUCP4A showed a protective role against mitochondrial dysfunction, by increasing mitochondrial membrane potential and ATP synthesis. To date, DmUCP4A is still an orphan of a biochemical function, although its possible involvement in mitochondrial uncoupling has been ruled out. This study shows that DmUCP4A expressed in bacteria and reconstituted in phospholipid vesicles catalyzes a unidirectional transport of aspartate, which is saturable and inhibited by mercurials and other mitochondrial carrier inhibitors to various degrees. Swelling experiments carried out in yeast mitochondria have demonstrated that the unidirectional transport of aspartate catalyzed by DmUCP4 is not proton-coupled. The biochemical function of DmUCP4A has been further confirmed in a yeast cell model, in which growth has required an efflux of aspartate from mitochondria. Notably, DmUCP4A is the first UCP4 homolog from any species to be biochemically characterized. In Drosophila melanogaster, DmUCP4A could be involved in the transport of aspartate from mitochondria to the cytosol, in which it could be used for protein and nucleotide synthesis, as well as in the biosynthesis of β-alanine and N-acetylaspartate, which play key roles in signal transmission in the central nervous system (Lunetti, 2022).
Lipid storage in fat tissue is important for energy homeostasis and cellular functions. Through RNAi screening in Drosophila fat body, this study found that knockdown of a Drosophila NAD kinase (NADK), which phosphorylates NAD to synthesize NADP de novo, causes lipid storage defects. NADK sustains lipogenesis by maintaining the pool of NADPH. Promoting NADPH production rescues the lipid storage defect in the fat body of NADK RNAi animals. Furthermore, NADK and fatty acid synthase 1 (FASN1) regulate mitochondrial mass and function by altering the levels of acetyl-CoA and fatty acids. Reducing the level of acetyl-CoA or increasing the synthesis of cardiolipin (CL), a mitochondrion-specific phospholipid, partially rescues the mitochondrial defects of NADK RNAi. Therefore, NADK- and FASN1-mediated fatty acid synthesis coordinates lipid storage and mitochondrial function (Xu, 2021).
Lipid homeostasis is important for human health, and its dysregulation is tightly associated with many metabolic diseases, such as type 2 diabetes, hepatic steatosis, cardiovascular disease, and cancer. Cellular lipid homeostasis is regulated by the opposing actions of lipid accumulation, including lipid uptake, de novo lipogenesis and lipid storage, and lipid mobilization, such as lipolysis, lipid oxidation, and lipid efflux. Excess lipid storage or insufficient lipid storage causes obesity or lipodystrophy, respectively (Xu, 2021).
Acetyl-CoA carboxylase (ACC) and FASN mediate fatty acid synthesis from acetyl-CoA during de novo lipogenesis. The fatty acids are then esterified for storage as neutral lipids such as triglycerides (TAGs). The lipid droplet, an organelle with a neutral lipid core and a phospholipid monolayer, is the hub for lipid storage. Understanding of the regulation of lipid storage and lipid droplet dynamics has significantly advanced in recent years. Many processes, including neutral lipid synthesis and degradation, composition of phospholipids, lipid droplet biogenesis and fusion, calcium homeostasis, and lipophagy, together determine lipid storage. Nevertheless, the mechanisms regulating lipid storage and lipid droplet dynamics in vivo are not completely clear (Xu, 2021).
To reduce lipid storage, TAG is mobilized through cytosolic lipolysis to release fatty acids, which are subsequently broken down, mainly in mitochondria, into acetyl-CoA units by lipid oxidation. Therefore, defective mitochondria often lead to lipid accumulation. For example, inhibition of β-oxidation in mitochondria causes lipid accumulation in Drosophila brain. Interestingly, besides conducting fatty acid oxidation, mitochondria also provide substrates and energy for de novo fatty acid synthesis. Both the acetyl-CoA and ATP required by fatty acid synthesis are derived from mitochondria. Impairment of mitochondrial function affects lipogenesis and lipid droplet accumulation. Therefore, impairment of mitochondrial function probably has a context-dependent effect on lipid storage (Xu, 2021).
Conversely, dysregulation of lipid storage also affects mitochondrial function. In the heart, cytoplasmic adipose TAG lipase (ATGL), which hydrolyzes TAG from lipid droplets, affects lipid storage and mitochondrial biogenesis and oxidative metabolism. Similarly, in islet β cells, ATGL knockdown impairs mitochondrial respiration and ATP production, and a PPARδ agonist rescues these mitochondrial defects (Xu, 2021).
Mechanistically, ATGL-mediated lipid droplet lipolysis induces the expression of genes involved in mitochondrial oxidation and respiration by activating the master regulators PPARα/PPARγ and PGC-1α. These studies pinpoint a close relationship between the mitochondrion and the lipid droplet, despite the compartmentalized features of lipid storage and lipid breakdown. Several metabolites, including acetyl-CoA and fatty acids, appear to mediate the two-way communication between these two organelles. De novo lipogenesis is tightly associated with acetyl-CoA and fatty acids. However, despite a few reports showing that lipogenesis inhibitors cause various mitochondrial dysfunctions in cancer, the question of whether and how de novo lipogenesis affects mitochondrial function has not been properly addressed (Xu, 2021).
Through an RNAi screen in Drosophila, this study found that CG6145, a cytosolic NAD kinase (NADK), affects lipid storage in fat body by providing NADPH, an essential reductant in lipogenesis. NADK RNAi causes similar de novo lipogenesis defects as FASN1 RNAi. More importantly, both NADK RNAi and FASN1 RNAi larvae exhibit reduced mitochondrial content. Finally, it was revealed that de novo fatty acid synthesis regulates mitochondrial mass, at least partially, by controlling PGC-1α acetylation and cardiolipin (CL) synthesis (Xu, 2021).
This study shows that NADK affects lipid storage and mitochondrial metabolism in Drosophila. NADK is essential for generating NADP and NADPH, the latter of which is important for de novo fatty acid synthesis. Besides lipid storage, NADK-mediated fatty acid synthesis also contributes to mitochondrial function, possibly through two different mechanisms: one is through acetyl-CoA and PGC-1α acetylation, and the other is through synthesis of the mitochondrion-specific phospholipid CL (Xu, 2021).
Despite the obvious requirement for NADPH in de novo fatty acid synthesis and other metabolic reactions, knowledge about the physiological function and impact of NADK on metabolic homeostasis in different organisms and tissues is limited. This study demonstrated the importance of NADK in animal lipid storage in vivo. NADK determines the level of NADP(H). Increasing NADPH availability rescues the defects in NADK RNAi, which confirms that NADPH is a key determinant of lipid storage. In support of this idea, NADPH-producing enzymes, such as G6PD and ME, promote lipid production in oleaginous microbes. The expression and activities of these enzymes are also correlated with lipid storage in mammals. These observations suggest that NADK and the level of NADPH are previously unappreciated regulators of organismal lipid storage. Interestingly, insulin, which promotes the synthesis and storage of lipids, activates NADK by Akt-mediated phosphorylation, which suggests that NADK may respond to physiological conditions to regulate lipid storage (Xu, 2021).
Besides lipogenesis, this stufy found that NADK also influences mitochondrial metabolism. The amounts of mitochondria and lipid droplets are decreased in both NADK RNAi and FASN1 RNAi, raising the possibility that these two closely linked organelles are co-regulated. Mitochondria regulate lipid metabolism by providing energy and substrates for lipogenesis and a site for fatty acid degradation. Lipid droplets, acting as an important organelle of lipid metabolism, also regulate mitochondrial function. Interestingly, elevating lipolysis by ATGL overexpression reduces the amount of lipid droplets, but it increases mitochondrial content, which suggests that reduced lipid storage per se is not the cause of the reduced mitochondrial mass in both NADK RNAi and FASN1 RNAi. Previous studies have shown that ATGL-mediated lipolysis promotes mitochondrial metabolism and biogenesis through activation of PPARs or Sirt1/PGC-1α. NADK RNAi and FASN1 RNAi exert a stronger effect on mitochondrial function than on lipolysis, which might be attributed to the severe decline in the level of fatty acids. Interestingly, PGC-1α acetylation mediates the regulation of mitochondrial function by both lipolysis and lipogenesis. Therefore, de novo fatty acid synthesis regulates the dynamics of both lipid droplets and mitochondria (Xu, 2021).
Fatty-acid-dependent activation of PPARs and Sirt1 is rather specific. The ligands of PPARs are primarily unsaturated and long-chain fatty acids, while Sirt1 is activated by monounsaturated fatty acids within a restricted range of concentrations. This study found that RNAi of the fat-body-specific PGC-1α homolog srl only moderately reduced mitochondrial mass, in contrast to the strong effect of NADK and FASN1 RNAi. In addition, knockdown of PPAR homologs in fat body caused no obvious mitochondrial phenotype. Therefore, it is likely that fatty acids also regulate mitochondrial function through other mechanism(s). In addition, the rescue of NADK RNAi and FASN1 RNAi by different exogenously supplied fatty acids (including saturated, monounsaturated, and odd-chain fatty acids) and by BMM overexpression suggests a general mechanism with limited or low fatty acid selectivity (Xu, 2021).
Phospholipid synthesis, which affects mitochondrial function in many ways, also requires fatty acids. CL is a mitochondrion-specific phospholipid and is important for almost every aspect of mitochondrial integrity, including crista organization, mitochondrial protein import, and assembly. It is a rather unique phospholipid, harboring four fatty acyl chains, and it undergoes remodeling, which makes it sensitive to the availability and composition of fatty acids. Importantly, the rescue of mitochondrial defects in NADK RNAi and FASN1 RNAi by several genetic manipulations to increase CL production suggests that decreased CL synthesis contributes to the mitochondrial phenotype in NADK RNAi and FASN1 RNAi. The mitochondrial morphology in NADK RNAi and FASN1 RNAi is not completely identical with CLS RNAi. In addition, the rescue effect of CLS overexpression is not comparable with fatty acid supplementation. These observations suggest that fatty acids might also regulate mitochondria through other mechanisms (Xu, 2021).
De novo fatty acid synthesis is important for many biological processes. For example, the activity of fatty acid synthesis is stimulated in some cancer cells or proliferating stem cells. Its inhibition suppresses cell proliferation and survival. It is generally thought that fatty acid synthesis mainly affects these processes by providing structural and signaling lipids\, and limited attention has been paid to the causative role of mitochondrial dysfunction, which is also important for cancer progression and stem cell homeostasis. For example, inhibition of PGC-1α or OXPHOS suppresses cancer cell survival and metastasis under oxidative or bioenergetic stress conditions. In addition, mitochondrial mass is associated with prostate cancer progression. Inhibition of mitochondrial biogenesis was identified as a therapeutic strategy for acute myeloid leukemia. Although OXPHOS activity is restricted in many cancer cells, mitochondrial content, dynamics, and metabolic activity are important for tumorigenesis and stem cell homeostasis (Xu, 2021).
Considering the findings of this study, it is possible that fatty acid synthesis-regulated mitochondrial function may be critical for cancer cell growth and stem cell differentiation. For example, fatty acid and lipid synthesis promote hepatocellular carcinoma development, accompanied by increased CL levels and OXPHOS activity. Inhibition of FASN or ACC reduces mitochondrial oxygen consumption, changes mitochondrial morphology, and affects the levels of mitochondrial proteins and metabolites in cancer and stem cells (Xu, 2021).
Both NADK and FASN are considered as potential targets for cancer therapy because of their lipogenic and other functions. NADK and FASN act as important regulators of lipid storage by restricting the capacity of fatty acid synthesis. This study showed that NADK- and FASN1-mediated fatty acid synthesis regulates mitochondrial function, probably by altering the levels of acetyl-CoA and CL. More physiological functions and molecular mechanisms of NADK and fatty acid synthesis may be revealed through the fatty-acid-mitochondrion link (Xu, 2021).
This study has demonstrated that increased PGC-1 acetylation and reduced CL synthesis are responsible for mitochondrial phenotype in NADK RNAi and FASN1 RNAi. However, reduced acetyl-CoA level and CLS overexpression only partially rescued mitochondrial phenotype. Exogenous fatty acid supplement completely restored mitochondrial mass in NADK RNAi and FASN1 RNAi, suggesting that fatty acid synthesis might regulate mitochondrial mass via other mechanisms as well. In addition, these studies were conducted in fat cells, which are specialized for lipid storage. It remains to be determined whether these findings apply to other cell types (Xu, 2021).
Respiratory complex I powers ATP synthesis by oxidative phosphorylation, exploiting the energy from NADH oxidation by ubiquinone to drive protons across an energy-transducing membrane. Drosophila melanogaster is a candidate model organism for complex I due to its high evolutionary conservation with the mammalian enzyme, well-developed genetic toolkit, and complex physiology for studies in specific cell types and tissues. Complex I was isolated from Drosophila, and its structure was determined, revealing a 43-subunit assembly with high structural homology to its 45-subunit mammalian counterpart, including a hitherto unknown homologue to subunit NDUFA3. The major conformational state of the Drosophila enzyme is the mammalian-type 'ready-to-go' active resting state, with a fully ordered and enclosed ubiquinone-binding site, but a subtly altered global conformation related to changes in subunit ND6. The mammalian-type 'deactive' pronounced resting state is not observed: in two minor states the ubiquinone-binding site is unchanged, but a deactive-type p-bulge is present in ND6-TMH3. This detailed structural knowledge of Drosophila complex I provides a foundation for new approaches to disentangle mechanisms of complex I catalysis and regulation in bioenergetics and physiology (Agip, 2023).
Mitochondrial complex I (NADH:ubiquinone oxidoreductase) is a crucial enzyme in cellular metabolism, central to NAD+ homeostasis, respiration, and oxidative phosphorylation, and a key contributor to the production of cellular reactive oxygen species (ROS). By catalyzing NADH oxidation in the mitochondrial matrix coupled to ubiquinone reduction in the inner membrane, it regenerates the oxidised NAD+ pool to sustain crucial metabolic processes, including the tricarboxylic acid cycle and β-oxidation, and provides reducing equivalents to the downstream complexes of the electron transport chain. The energy from NADH:ubiquinone oxidoreduction is harnessed to transport four protons across the inner membrane, supporting the proton motive force (Δp) that drives ATP synthesis and transport processes. These central roles of complex I in both metabolism and oxidative stress make complex I dysfunctions, induced by genetic, pharmacological, and environmental factors, some of the most frequent primary causes of mitochondrial diseases, as well as a contributor to many socially and economically important diseases common in ageing populations. For example, ROS production by complex I operating in reverse, during 'reverse electron transfer' (RET, Δp-driven ubiquinol:NAD+ oxidoreduction), is a major contributor to the tissue damage that occurs in strokes and heart attacks, during ischaemia-reperfusion (IR) injury (Agip, 2023).
Mammalian complex I is a 1 MDa asymmetric assembly of 45 subunits, encoded on both the nuclear and mitochondrial genomes. Fourteen of them (seven nuclear and seven mitochondrial) are the core subunits conserved in all complex I homologues that are essential for catalysis, whereas the other 31 subunits are supernumerary subunits that are involved in enzyme assembly, stability, and regulation, or that have independent roles within the cell. Bioinformatic analyses have indicated how the cohort of supernumerary subunits has been augmented gradually throughout the evolution of the eukaryotic complex, and an increasing range of structural analyses of different species of complex I now illustrates the diversity of the supernumerary subunit cohorts that have developed in different eukaryotic lineages (Agip, 2023).
For mammalian complex I, the form of the enzyme most relevant in medicine, single-particle electron cryomicroscopy (cryo-EM) has yielded detailed structural information on multiple different states of the complex. However, detailed structure-function studies are limited for the mammalian enzyme due to substantial challenges in creating and studying genetic variants in representative mammalian model systems, such as mouse. Whereas simpler model systems, such as α-proteobacteria or yeast species, allow far greater opportunities for genetic studies, the protein compositions of their complex I vary substantially from the mammalian enzyme, fail to recapitulate key characteristics and behaviour of the mammalian complex such as the 'active/deactive transition', and the physiological environments in which the variant complexes can be studied are very restricted. Most relevant for this study, the active and deactive states of mammalian complex I are two biochemically and structurally characterised resting states of the complex: the 'active' ready-to-go resting state and the 'deactive' pronounced resting state. They differ both in their global conformations and in the status of local structural features. In particular, the ubiquinone-binding site in the active state is fully enclosed and sealed, whereas in the deactive state disorder in the enclosing loops opens the site to the matrix. The active and deactive resting states have also been referred to as the 'closed' and 'open' states of the mammalian enzyme on the basis of changes in the apparent angle between their membrane and hydrophilic domains. Finally, it is noted that there is currently substantial controversy about the biochemical and physiological relevance of the open states of the mammalian complex, which have recently been proposed to include not only the deactive resting state but also on-cycle catalytic intermediates (Agip, 2023).
The fruit fly, Drosophila melanogaster, is a powerful genetically tractable model organism for metazoa. Drosophila encodes a complex I with a composition that closely resembles that of the mammalian complex, with clear homologues to 42 of the 44 mammalian subunits identified. Therefore, in addition to providing an additional model system for studying the mechanism of complex I catalysis (also accessible in simpler unicellular models), variants in Drosophila complex I can be studied for their effects on regulation and assembly. Furthermore, Drosophila can potentially be exploited to investigate features of complex I function that are observed for mammalian complex I, but not universal features of the enzyme in simpler organisms, such as the active/deactive transition, RET, and the involvement of complex I in supercomplexes. For instance, studies in Drosophila have proposed that RET-ROS increase lifespan and Drosophila are remarkably resistant to hypoxic or anoxic exposure, which might provide insights into pathological mechanisms of RET-mediated IR injury. Furthermore, with substantial tissues, such as indirect flight muscles, highly enriched with mitochondria, Drosophila represent an attractive animal model for the analysis of basic mitochondrial biology, offering a complex physiological system for the generation and study of complex I genetic variants at the whole organism or tissue-specific level, as well as the involvement of complex I in differing physiological conditions (Agip, 2023).
To date, no detailed molecular studies of Drosophila complex I have been pursued to confirm its structural and functional similarity with the mammalian enzyme, or exploit its potential as a metazoan model system. Therefore, this study sought to structurally and biochemically evaluate Drosophila as a model system for mammalian complex I. Structures were determined for three distinct conformational states of the Drosophila enzyme and compared to well-characterised resting states of the mammalian complex, leading to new insights into the mammalian active/deactive transition and enhancing understanding of the conformational link between the ubiquinone-binding site and the proximal membrane domain. This study thus presents detailed knowledge of Drosophila complex I at the molecular level and confirm and define its relationships to the mammalian enzyme (Agip, 2023).
The structures determined in this study for Drosophila complex I confirm its close relationships with mammalian complex I and thus its potential as a powerful genetically tractable model system for studying mammalian-specific aspects of complex I biology. The close-to-identical subunit compositions and structures of the mammalian and Drosophila enzymes now enable genetic approaches to be applied to elucidate, for example, the roles of the supernumerary subunits, the assembly pathway, and the detrimental effects of clinically identified pathological point mutations. Importantly, these aspects can be studied in physiologically relevant in vivo environments and in specific cell types and tissues, extending the scope of earlier studies in cultured mammalian cells. However, the structures also reveal limitations in Drosophila as a model organism for complex I, as the Drosophila enzyme, despite its remarkable similarity to the mammalian enzyme, does not undergo the full mammalian-type active/deactive transition. Cryo-EM analyses revealed the major class of enzyme particle (Dm1) in the active resting state, with all the characteristics of the mammalian active state enzyme. Two minor states (Dm2 and Dm3) also more closely resemble the active state, and this study was unable to either detect a mammalian-type deactive resting state in biochemical assays, or to generate one by incubation of the enzyme at 37°C (the method used to deactivate mammalian complex I). However, it is noted that the biochemical assay relies on the availability of ND3-Cys41, just one characteristic that distinguishes the mammalian active and deactive states; the functional consequences of conversion to the Dm2 state are currently unknown, most notably whether it is (like the mammalian active state) able to catalyse RET, or (like the mammalian deactive state) unable to do so (Agip, 2023).
Comparison of the Dm1 (active) and Dm2 (twisted) structures of Drosophila complex I determined in this study suggests that the Dm2 state is a relaxed state, which may be considered a structurally curtailed form of the full mammalian-type deactive transition. In changes that also occur in the mammalian transition, a &pi&-bulge forms in ND6-TMH3, and the nearby sidechain of ND1-TMH4-Tyr149 flips in conformation. Although water molecules cannot be resolved in these structures, these changes are expected to alter the connectivity between the E-channel and the central axis of charged residues along the membrane domain as reported previously for mammalian, yeast, and bacterial species. Furthermore, the limited and local changes observed in the ND6 region result in a limited twisting of the global conformation in the Dm2 state, a motion that qualitatively resembles (but to a much lesser extent) the twisting of the deactive enzyme. However, the cascade of changes that also occurs in the mammalian-type deactive transition does not follow: ND1-TMH4 does not straighten its conformation, the trigonal junction between ND3-Cys41, NDUFS2-His93, and ND1-Tyr134 is preserved, and so the conformational change from ND6-TMH3 does not propagate to the ubiquinone-binding site, which remains fully ordered, sealed from the matrix, and in its active state (see Local structural elements show that the Dm2 state of Drosophila complex I most closely resembles the mammalian active state, with only two deactive-like features in the membrane domain.). This lack of direct correlation between the status of the α-helix/π-bulge and the structuring of the ubiquinone-binding site (see Schematic representation of the status of local active/deactive elements in the (a) Dm1 and (b) Dm2 states of Drosophila complex I. argues against a concerted and structurally enforced connection between them being crucial for catalysis. Although structures of complex I from other (non-mammalian) species have also been reported with a π-bulge in ND6-TMH3 but without the 'opening' of the ubiquinone-binding site observed in the mammalian deactive state, the Drosophila Dm2 structure is the first example in which the π-bulge and a fully ordered, active ubiquinone-binding site have been observed together. These structures are consistent with the elements that change during the mammalian deactive transition (such as the π-bulge) being mobile during catalysis, but do not suggest that they move in a coherent and coordinated transition, with the ubiquinone-binding site open to the matrix, during catalysis (Agip, 2023).
The observation, together, of the active Dm1 state and the 'curtailed-deactive' Dm2 state raises two questions: what causes the π-bulge to form in Dm2, and why does the conformational cascade to the mammalian-type deactive state not occur in the Drosophila enzyme? First, it is possible that delipidation of the complex during detergent extraction removes the intercalated phospholipid that obstructs π-bulge formation in the Dm1 state, allowing conversion to Dm2. However, a similar intercalated phospholipid has not been observed in any mammalian active-state structure, so it may only bind when catalysis stops, or be an artefact of enzyme purification. Indeed, if ND6-TMH3 converts between its π-bulge and α-helical structures during catalysis, then the intercalating phospholipid is very unlikely to be present in the α-helical state, moving repeatedly in and out. Alternatively, it is possible that enzyme twisting, induced by loss of the NDUFS4 tether from the NDUFA5/NDUFA10 interface during purification, causes the π-bulge to form: this possibility may be addressed in future by genetic truncation of the NDUFS4 tether from the N-terminus of the mature subunit. Second, if formation of the π-bulge in Drosophila represents a curtailed-deactive transition, then conversion to a full mammalian-type deactive state would be accompanied by further twisting, disruption of the NDUFA5/NDUFA10 interface, and destructuring of the ubiquinone-binding site. That these changes are not observed in Drosophila complex I is likely due to the modified domain disposition in the Dm1 state that is stabilised by the structure of the connecting subdomain and accommodating changes in linked structures such as the NDUFA5/NDUFA10 interface. It is proposed that the stable domain disposition is resistant to further twisting and so resists the local changes that accompany it in the mammalian deactive transition. Computational simulations of the Dm1 structure may help further elucidate the answer to this question in future. Notably, the current proposal implies high activation energy barriers for the 'opening' of the ubiquinone-binding site to the matrix in the Drosophila enzyme, arguing against opening and closing of the site during catalysis (Agip, 2023).
The Dm3 'cracked' state is not discussed in detail as it is suspected to be an artefact resulting from detergent-induced loss of stability in the distal membrane domain of the Dm2 state. Similar opening and relaxation of the ND2-ND4 interface has also been observed in the 'slack' state of bovine complex I, as well as in a catalytically inactive state of complex I from rhesus macaque, and in pronounced open states of the ovine complex. In all cases, opening of the ND2-ND4 interface is linked to loss of density for nearby subunit NDUFA11, and to changes in the C-terminal section of the ND5 transverse helix and anchor helix. It may result from delipidation during enzyme purification, most likely removal of phospholipids from the interface on both sides of the complex, including 'behind' the transverse helix. Consistent with this picture, treatment of the mammalian enzyme with zwitterionic detergents or prolonged incubation in detergent solution leads to fractionation at this interface (Agip, 2023).
The deactive transition and RET are linked in mammalian complex I biology, as deactivation protects against the burst of ROS production that occurs upon reperfusion by RET (RET-ROS), driven by oxidation of the reduced succinate pool that accumulates during ischaemia, leading to IR injury. The deactivation of complex I minimises the RET-ROS burst and tissue damage upon reperfusion because the deactive state of mammalian complex I is unable to catalyse RET. An elegant demonstration is provided by the ND6-P25L variant of mouse complex I, which deactivates much more rapidly than the wild-type enzyme, preventing RET-ROS catalysis and thereby protecting against IR injury. Strikingly, while Drosophila do not appear to adopt a mammalian-type deactive state, they are able to survive long periods of hypoxia followed by reoxygenation, raising the question of whether they are protected by a corresponding mechanism. ROS production by RET has been described in studies of Drosophila mitochondria (although not demonstrated directly in the isolated enzyme), and the ability of Drosophila complex I to catalyse RET is consistent with it persisting in the active state (Dm1) when catalysis stops, rather than deactivating. Alternative mechanisms are therefore required to explain the resistance of Drosophila to hypoxia−reoxygenation challenges, such as greater robustness to oxidative stress from a RET-ROS-induced stress-responsive transcriptional programme and/or metabolic adaptations . Future genetic studies that exploit structural insights will illuminate these mechanisms and provide new perspectives on the mechanisms of mammalian complex I (Agip, 2023).
Mitochondria are optically responsive organelles producing energy for cell function via adenosine triphosphate (ATP). But ATP production appears to vary over the day. This study used Drosophila melanogaster to reveal daily shifts in whole animal ATP production in a tight 24 hours' time series. A marked production peak in the morning was shown to declines around midday and remains low through afternoon and night. ATP production can be improved with long wavelengths (>660 nm), but apparently not at all times. Hence, flies were treated with 670 nm light to reveal optimum times. Exposures at 670 nm resulted in a significant ATP increases and a shift in the ATP/adenosine diphosphate (ADP) ratio at 8.00 and 11.00, whilst application at other time points had no effect. Hence, light-induced ATP increases appear limited to periods when natural production is high. In summary, long wavelength influences on mitochondria are conserved across species from fly to human. Determining times for their administration to improve function in ageing and disease are of key importance. This study progresses this problem (Shinhmar, 2022).
The mitochondrial electron transport chain (mETC) contains molecular targets of volatile general anesthetics (VGAs), which places carriers of mutations at risk for anesthetic complications. The ND-2360114 and mt:ND2del1 lines of fruit flies (Drosophila melanogaster) that carry mutations in core subunits of Complex I of the mETC replicate numerous characteristics of Leigh syndrome (LS) caused by orthologous mutations in mammals and serve as models of LS. ND-2360114 flies are behaviorally hypersensitive to volatile anesthetic ethers and develop an age- and oxygen-dependent anesthetic-induced neurotoxicity (AiN) phenotype after exposure to isoflurane but not to the related anesthetic sevoflurane. The goal of this paper was to investigate whether the alkane volatile anesthetic halothane and other mutations in Complex I and in Complexes II-V of the mETC cause AiN. It was found that (1) ND-2360114 and mt:ND2del1 were susceptible to toxicity from halothane; (2) in wild-type flies, halothane was toxic under anoxic conditions; (3) alleles of accessory subunits of Complex I predisposed to AiN; and (iv) mutations in Complexes II-V did not result in an AiN phenotype. It is concluded that AiN is neither limited to ether anesthetics nor exclusive to mutations in core subunits of Complex I (Borchardt, 2023).
In most eukaryotic cells, fatty acid synthesis (FAS) occurs in the cytoplasm and in mitochondria. However, the relative contribution of mitochondrial FAS (mtFAS) to the cellular lipidome is not well defined. This study shows that loss of function of Drosophila mitochondrial enoyl coenzyme A reductase (Mecr), which is the enzyme required for the last step of mtFAS, causes lethality, while neuronal loss of Mecr leads to progressive neurodegeneration. A defect in Fe-S cluster biogenesis and increased iron levels were observed in flies lacking mecr, leading to elevated ceramide levels. Reducing the levels of either iron or ceramide suppresses the neurodegenerative phenotypes, indicating an interplay between ceramide and iron
metabolism. Mutations in human MECR cause pediatric-onset neurodegeneration, and this study shows that human-derived fibroblasts display similar elevated ceramide levels and impaired iron homeostasis. In summary, this study identifies a role of mecr/MECR in ceramide and iron metabolism, providing a mechanistic link between mtFAS and neurodegeneration (Dutta, 2023).
Complex I (CI) deficiency in mitochondrial oxidative phosphorylation (OXPHOS) is the most common cause of mitochondrial diseases, and limited evidence-based treatment options exist. Although CI provides the most electrons to OXPHOS, complex II (CII) is another entry point of electrons. Enhancement of this pathway may compensate for a loss of CI; however, the effects of boosting CII activity on CI deficiency are unclear at the animal level. 5-Aminolevulinic acid (5-ALA) is a crucial precursor of heme, which is essential for CII, complex III, complex IV (CIV) and cytochrome c activities. This study shows that feeding a combination of 5-ALA hydrochloride and sodium ferrous citrate (5-ALA-HCl + SFC) increases ATP production and suppresses defective phenotypes in Drosophila with CI deficiency. Knockdown of sicily, a Drosophila homolog of the critical CI assembly protein NDUFAF6, caused CI deficiency, accumulation of lactate and pyruvate and detrimental phenotypes such as abnormal neuromuscular junction development, locomotor dysfunctions and premature death. 5-ALA-HCl + SFC feeding increased ATP levels without recovery of CI activity. The activities of CII and CIV were upregulated, and accumulation of lactate and pyruvate was suppressed. 5-ALA-HCl + SFC feeding improved neuromuscular junction development and locomotor functions in sicily-knockdown flies. These results suggest that 5-ALA-HCl + SFC shifts
metabolic programs to cope with CI deficiency. Bullet outline 5-Aminolevulinic acid (5-ALA-HCl + SFC) increases ATP production in flies with complex I deficiency.5-ALA-HCl + SFC increases the activities of complexes II and IV.5-ALA-HCl + SFC corrects metabolic abnormalities and suppresses the detrimental phenotypes caused by complex I deficiency (Nozawa, 2023).
Several phospholipid (PL) molecules are intertwined with some mitochondrial complex I (CI) subunits in the membrane domain of CI, but their function is unclear. This study reports that when the Drosophila melanogaster ortholog of the intramitochondrial PL transporter, STARD7 (CG6565), is severely disrupted, assembly of the oxidative phosphorylation (OXPHOS) system is impaired, and the biogenesis of several CI subcomplexes is hampered. However, intriguingly, a restrained knockdown of STARD7 impairs the incorporation of NDUFS5 and NDUFA1 into the proximal part of the CI membrane domain without directly affecting the incorporation of subunits in the distal part of the membrane domain, OXPHOS complexes already assembled, or mitochondrial cristae integrity. Importantly, the restrained knockdown of STARD7 appears to induce a modest amount of cardiolipin remodeling, indicating that there could be some alteration in the composition of the mitochondrial phospholipidome. It is concluded that PLs can regulate CI biogenesis independent of their role in maintaining mitochondrial membrane integrity (Murari, 2023).
Lung adenocarcinoma (LUAD) has high morbidity and is prone to recurrence. TIMELESS (TIM), which regulates circadian rhythms in Drosophila, is highly expressed in various tumors. Tumor samples from patients with LUAD patient data from public databases were used to confirm the relationship of TIM expression with lung cancer. LUAD cell lines were used and siRNA of TIM was adopted to knock down TIM expression in LUAD cells, and further cell proliferation, migration and colony formation were analyzed. By using Western blot and qPCR, the influence was detected of TIM on epidermal growth factor receptor (EGFR), sphingosine kinase 1 (SPHK1) and AMP-activated protein kinase (AMPK). With proteomics analysis, this study comprehensively inspected the different changed proteins influenced by TIM, and global bioinformatic analysis was performed. TIM expression was found to be elevated in LUAD and that this high expression was positively correlated with more advanced tumor pathological stages and shorter overall and disease-free survival. TIM knockdown inhibited EGFR activation and also AKT/mTOR phosphorylation. This study also clarified that TIM regulated the activation of SPHK1 in LUAD cells. And with SPHK1 siRNA to knock down the expression level of SPHK1, it was found that EGFR activation were inhibited greatly too. Quantitative proteomics techniques combined with bioinformatics analysis clarified the global molecular mechanisms regulated by TIM in LUAD. The results of proteomics suggested that mitochondrial translation elongation and termination were altered, which were closely related to the process of mitochondrial oxidative phosphorylation. It was further confirmed that TIM knockdown reduced ATP content and promoted AMPK activation in LUAD cells. This study revealed that siTIM could inhibit EGFR activation through activating AMPK and inhibiting SPHK1 expression, as well as influencing mitochondrial function and altering the ATP level; TIM's high expression in LUAD is an important factor and a potential key target in LUAD (Yin, 2023).
Respiratory complexes and cardiolipins have exceptionally long lifetimes. The fact that they co-localize in mitochondrial cristae raises the question of whether their longevities have a common cause and whether the longevity of OXPHOS proteins is dependent on cardiolipin. To address these questions, a method was developed to measure side-by-side the half-lives of proteins and lipids in wild-type Drosophila and cardiolipin-deficient mutants. Adult flies were fed with stable isotope-labeled precursors ((13)C(6)(15)N(2)-lysine or (13)C(6)-glucose), and the relative abundance of heavy isotopomers in protein and lipid species was determined by mass spectrometry. To minimize the confounding effects of tissue regeneration, this analysis was restricted to the thorax, the bulk of which consists of post-mitotic flight muscles. Analysis of 680 protein and 45 lipid species showed that the subunits of respiratory complexes I-V and the carriers for phosphate and ADP/ATP were among the longest-lived proteins (average half-life of 48 ± 16 days) while the molecular species of cardiolipin were the longest-lived lipids (average half-life of 27 ± 6 days). The remarkable longevity of these crista residents was not shared by all mitochondrial proteins, especially not by those residing in the matrix and the inner boundary membrane. Ablation of cardiolipin synthase, which causes replacement of cardiolipin by phosphatidylglycerol, and ablation of tafazzin, which causes partial replacement of cardiolipin by monolyso-cardiolipin, decreased the lifetimes of the respiratory complexes. Ablation of tafazzin also decreased the lifetimes of the remaining cardiolipin species. These data suggest that an important function of cardiolipin in mitochondria is to protect respiratory complexes from degradation (Rem, 2023).
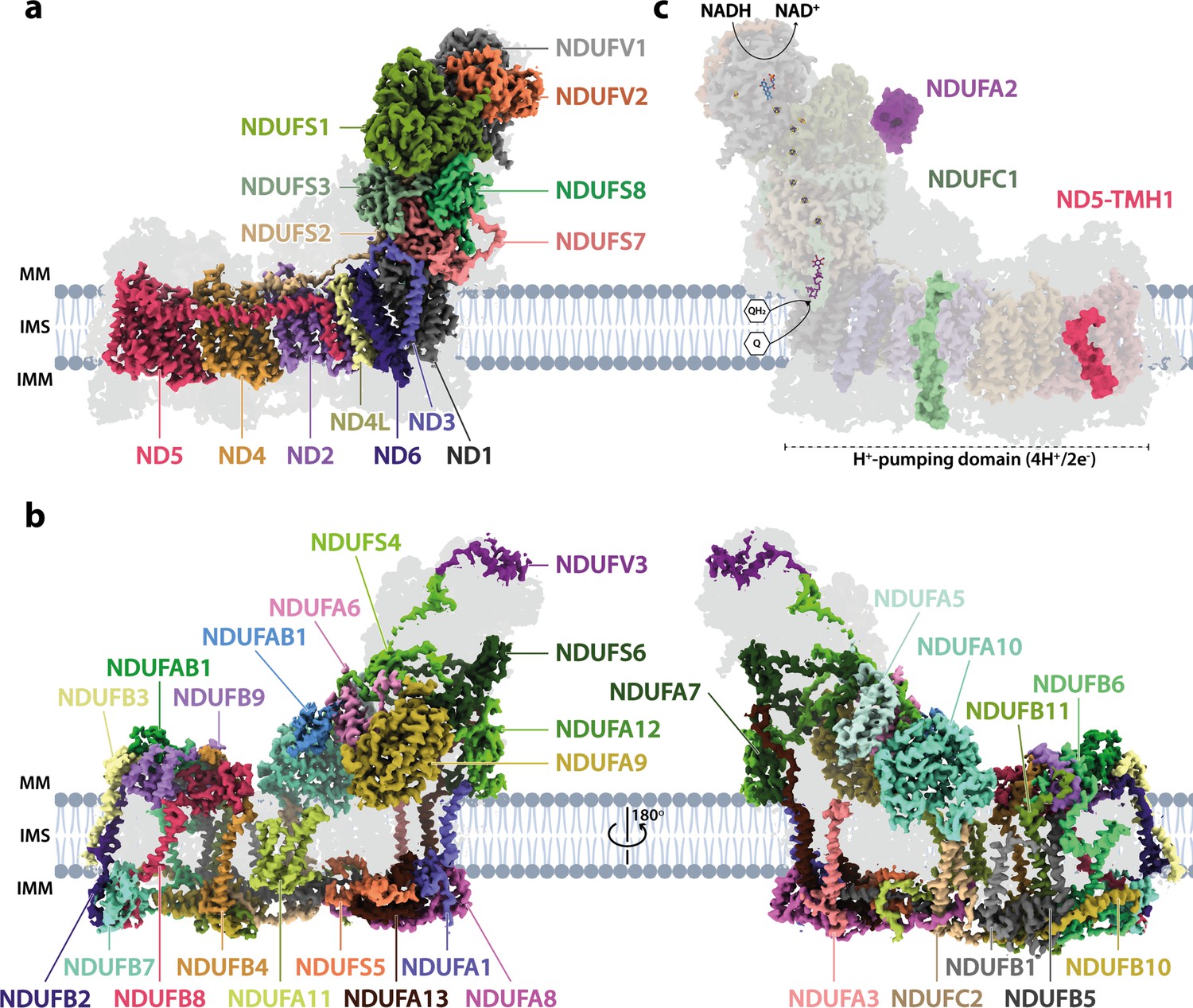{kind=link}
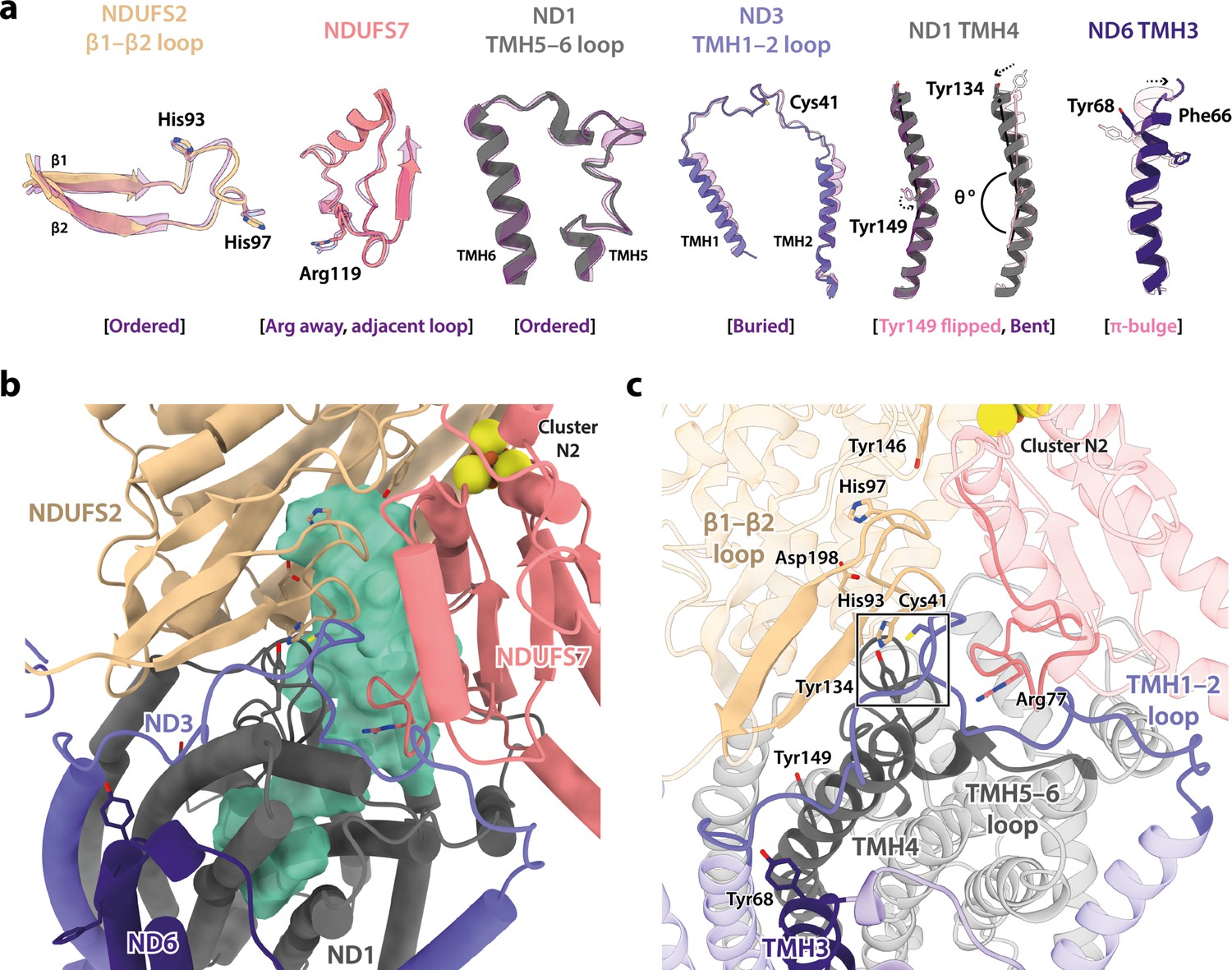{kind=link}